Locations:

Case report illustrates the importance of maintaining high levels of clinical suspicion
By Vinni Makin, MD, and Yumiko Tsushima, MD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
A 54-year-old man presented to the emergency department with new onset oral thrush and right-sided facial swelling after dental work. He presented to his dermatologist eight months previously with complaints of darkening of the skin and photosensitivity. An elevated adrenocorticotropic hormone (ACTH) was noted at this time and subsequent testing with 24-hour urinary cortisol showed mild elevation (Table 1). The patient was referred to Endocrinology, but failed to follow-up. One month later, following a motor vehicle accident, the patient had a chest x-ray that incidentally revealed a mediastinal mass. Computed tomography (CT) of the chest confirmed an anterior mediastinal mass measuring 10.7 x 6.3 x 11.0 cm. A biopsy at an outside hospital demonstrated a benign lipoma.
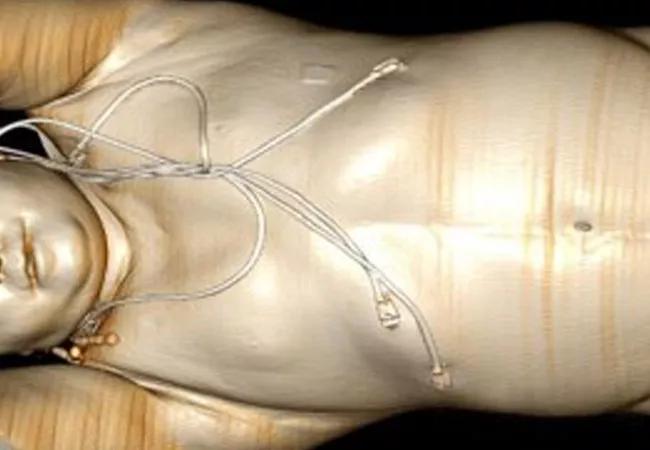
When the patient was seen by Endocrinology during the present hospital admission, he reported proximal muscle weakness and central weight gain of 25 pounds. Physical exam was notable for a cushingoid appearance, facial fullness and plethora (Figure 1A), dorsocervical and supraclavicular fat pads with hyperpigmentation of the sun-exposed areas. Labs revealed severe hypokalemia resistant to treatment, metabolic alkalosis, rhabdomyolysis and leukocytosis without fever. Serum random cortisol and ACTH were elevated and cortisol was not suppressed with 8mg dexamethasone. Whole body PET-CT demonstrated a mild to moderate FDG avid anterior mediastinal mass, as well as symmetric thickening and hypermetabolism of the adrenal glands, likely due to adrenal hyperplasia. MRI of the pituitary noted normal size and configuration of the pituitary gland without evidence of adenoma or suprasellar mass.
Advertisement
Table 1: Pertinent laboratory findings
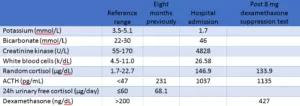
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/f8fd1f55-cd27-4408-8482-290be59475aa/table-300x106_png)
The patient’s symptoms and biochemical evidence of overwhelming hypercortisolism were concerning for ectopic Cushing’s syndrome given the undeniable presence of a large mediastinal mass. However, the previous diagnosis of a benign lipoma complicated the clinical picture. This prompted re-examination of CT chest images from the outside hospital, which revealed a mediastinal mass that lacked fat attenuation (Figure 1B), findings inconsistent with a lipoma. From this realization, we were able to pursue management for suspected ectopic ACTH syndrome (EAS) secondary to the mediastinal mass.
The patient was started on ketoconazole and mifepristone as pre-surgical treatment of EAS and the hypokalemia and new onset hypertension and hyperglycemia were managed simultaneously. The patient underwent surgical resection of the mediastinal mass along with pericardial resection and wedge resection of the left upper lobe of the lung. Pathology of the anterior mediastinal mass revealed a 12.6 x 8.0 x 4.5cm thymic neuroendocrine carcinoma, stage T3N1M0 due to direct invasion into the lung and metastases to regional lymph nodes. Serum ACTH and cortisol both rapidly declined post-operatively and the patient was transitioned to oral hydrocortisone.
The patient has had regular follow-up as an outpatient. He had lost 27 pounds by the time of his 2-month follow-up, and the hypertension and hyperglycemia resolved. ACTH and cortisol levels have normalized. Hydrocortisone was tapered off with recovery of adrenal function.
Advertisement
The most common tumors of the anterior mediastinum are thymomas, germ cell tumors, lymphomas and thyroid tissue. NETs of the thymus are extremely rare, constituting less than 0.5% of all NETs and less than 5% of anterior mediastinal tumors. It is three times more common in men1. One third to half of thymic NETs are functional and CS is the most common associated endocrinopathy.2-3
Ectopic ACTH Syndrome (EAS) from thymic NETs has a poorer prognosis than non-functioning tumors, possibly due to the aggressive nature of the tumor resulting in severe metabolic derangements from CS.4 Patients present with rapid onset of hypercortisolic features, such as proximal muscle weakness, significant hypokalemia, metabolic alkalosis, treatment resistant hypertension, hyperglycemia and edema.5 Hypercortisolemia also increases patients’ risk of bacterial and opportunistic infections, as seen in our patient who presented with oral thrush.6 Overall survival for thymic NETs is poor and is reported to be 28% at five years and 10% at ten years.7
Cushing’s syndrome (CS) may be ACTH-dependent (pituitary-origin or ectopic) or ACTH-independent (adrenal-origin). EAS compromises approximately 10% of ACTH dependent CS8 and is most frequently associated with gastrointestinal carcinoid tumors or small cell lung carcinoma. The presentation of EAS may vary widely, which makes it a diagnostic challenge, but the severity of metabolic derangements caused by hypercortisolemia is life-threatening and must be addressed promptly.
Advertisement
In order to diagnose EAS, a high clinical suspicion together with biochemical evidence of endogenous hypercortisolism and elevated ACTH is required. The challenge lies in the difficulty of distinguishing between EAS and pituitary CS (otherwise known as Cushing’s disease) because localizing the ectopic tumor can be difficult. In a study from Isidori et al, the tumor was only identified in 65% of patients after initial investigation and the source was never found in 12.5% of the cases.
The initial step is to prove endogenous hypercortisolism (Figure 2).
Figure 2: Diagnosis of Cushing’s syndrome
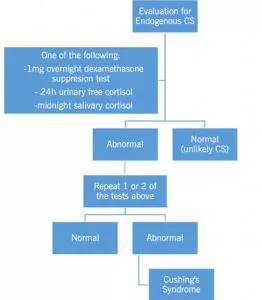
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/fe7985bc-e47e-49bc-9088-d0f0682239fd/19-EMI-1362-Hypercortisolism-CQD-Inset-2-262x300_jpg)
Adapted from Nieman et al.9
Then, the next step is to establish the cause of CS (Figure 3).
Figure 3: Establishing cause of CS
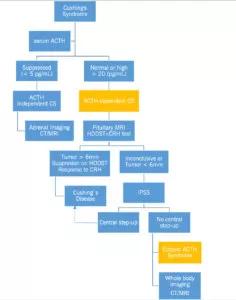
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/d0c192b6-c694-4e3c-82f8-c47c1e299f6e/19-EMI-1362-Hypercortisolism-CQD-Inset-3-236x300_jpg)
CRH=corticotropin-releasing hormone, HDDST=high dose dexamethasone suppression test, IPSS=inferior petrosal sinus sampling.
Adapted from Newell-Price et al.10
In order to differentiate between CD and EAS, the high dose dexamethasone suppression test (HDDST) is widely used. Serum cortisol suppressed to < 50% of baseline is suggestive of Cushing’s disease. Clinicians will sometimes perform a CRH stimulation test in addition to HDDST for further evaluation because of the variable sensitivity of the HDDST. There have been case series that have proposed that serum ACTH > 200 pg/ml suggests EAS over CD.11 IPSS was not performed in our case due to non-suppressible HDDST, negative pituitary MRI and high clinical suspicion of EAS.
Unfortunately, even with surgical resection of the tumor, recurrence remains high. Chemotherapy and radiation therapy are available for advanced disease and recurrences, but these adjuvant therapies do not result in statistically significant survival outcomes.
Advertisement
The diagnosis of CS from EAS remains challenging. In our case, the mediastial mass was mistakenly diagnosed as a benign lipoma due to sampling error. However, the key to a correct diagnosis was reviewing the imaging characteristics of the mass on CT and noticing that they were not consistent with a lipoma, and then combining this finding with the biochemical evidence for ACTH-dependent CS. We emphasize the importance of piecing together all aspects of the diagnostic evaluation and the need to maintain a high clinical suspicion for EAS in order to correctly diagnose and manage these patients.
Please note: The images related to this case were originally published by these authors here, and have been reused according to the Creative Commons license.
Advertisement

Pheochromocytoma case underscores the value in considering atypical presentations

Advocacy group underscores need for multidisciplinary expertise

A reconcilable divorce

A review of the latest evidence about purported side effects

High-volume surgery center can make a difference

Advancements in equipment and technology drive the use of HCL therapy for pregnant women with T1D

Patients spent less time in the hospital and no tumors were missed

A new study shows that an AI-enabled bundled system of sensors and coaching reduced A1C with fewer medications