Locations:

New drugs, diagnostic tools are starting to revolutionize care
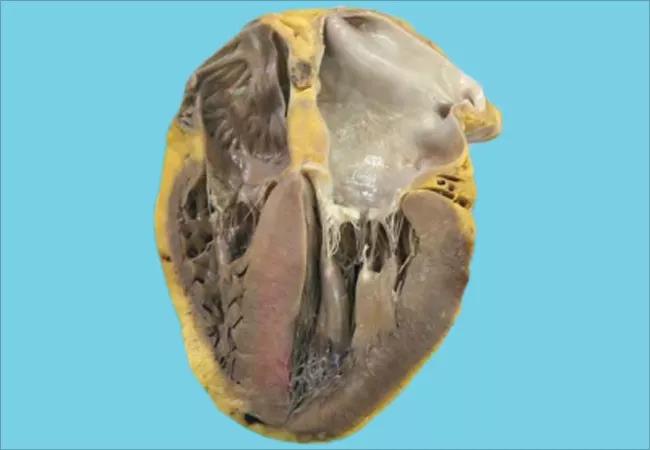
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/3508fff2-b0d9-49b5-8cd5-77bf82b88f7d/18-HRT-5612-cardiac-amyloidosis-650x450_jpg)
18-HRT-5612 cardiac-amyloidosis-650×450
Cardiac amyloidosis, traditionally considered a rare and dire disease, is receiving increasing attention as sophisticated diagnostics are identifying more patients and novel therapies are emerging to combat it.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Among the latest signs of progress are the encouraging results of the ATTR-ACT study of the drug tafamidis presented as a late-breaking trial at the European Society of Cardiology Congress in late August and simultaneously published online by the New England Journal of Medicine.
Tafamidis was studied for treating one of the two main types of cardiac amyloidosis — transthyretin (ATTR) amyloidosis, which is caused by misfolding of the liver-derived transthyretin protein, leading to amyloid fibril formation and deposition in the myocardium. Although both types cause cardiac amyloidosis, ATTR amyloidosis is a very distinct disease from light chain (AL) amyloidosis, in which plasma cells in the bone marrow produce an amyloidogenic light chain fragment. The pathophysiology, prognosis and treatment of the two types differ considerably.
Patients with AL amyloidosis are already benefiting from drugs developed to fight multiple myeloma. The triple drug strategy of bortezomib, cyclophosphamide and dexamethasone has significantly improved prognosis. A new drug, daratumumab, also shows promise for increasing survival in clinical trials.
In contrast, treatment for ATTR amyloidosis had been confined to symptom management and, in some patients, heart transplant. Until recently, there was no specific medical treatment for this type of amyloidosis, which can be hereditary, stemming from any of more than 120 identified genetic mutations of the transthyretin protein, or wild-type (formerly called senile cardiac amyloidosis), a disease of aging wherein there is no mutation. The disease usually presents as a neuropathy or cardiomyopathy (or a mixed phenotype).
Advertisement
Despite the long dearth of therapies for ATTR amyloidosis, recent clinical trials offer high hopes, and two new FDA-approved drugs have just come to market to treat the hereditary variant of ATTR amyloidosis.
Not yet approved but proven to be effective is the above-mentioned tafamidis, a small molecule that binds to transthyretin and prevents misfolding. The newly published phase 3 ATTR-ACT trial involved 441 patients with ATTR amyloidosis at 48 sites in 13 countries. Importantly, the majority of patients — 74 percent — had wild-type ATTR. Tafamidis significantly reduced all-cause mortality at 30 months relative to placebo (30 percent vs. 43 percent) and resulted in fewer cardiovascular-related hospitalizations. Treated patients also had a slower rate of decline in the 6-minute walk test and overall health as measured by the Kansas City Cardiomyopathy Questionnaire. Adverse events in the treatment and placebo arms were comparable.
“The degree of survival benefit from tafamidis was impressive,” says Cleveland Clinic cardiologist Mazen Hanna, MD, lead investigator of the ATTR-ACT trial at Cleveland Clinic and a co-author of the published study. “It will change the landscape of this disease and offers long-awaited hope to our patients with wild-type TTR cardiac amyloidosis in addition to those with the hereditary variant.”
Dr. Hanna explains that tafamidis slows rather than reverses disease, underscoring the urgency of the need for diagnosing and treating ATTR amyloidosis as early as possible. He is hopeful the drug will be approved by the FDA next year, as “many patients are waiting anxiously to get it.” (Watch this 4-minute video of Dr. Hanna recapping the pivotal ATTR-ACT study.)
Advertisement
In August 2018, patisiran (Onpattro™) became the first FDA-approved drug for the treatment of polyneuropathy associated with hereditary ATTR amyloidosis. An RNA inhibitor, it works by blocking production of the transthyretin protein. While clinical trials have raised expectations for patisiran’s use against ATTR cardiac amyloidosis, the drug’s current price tag — estimated at about $450,000 per year — may be challenging for many patients.
Another RNA inhibitor, inotersen (Tegsedi™), was approved by the FDA in early October for the treatment of hereditary ATTR amyloidosis polyneuropathy and will add another option for patients.
Dr. Hanna notes that other promising therapies in clinical trials include AG10, which works like tafamidis, and two “fibril disruptors” — PRX004 and anti-SAP antibodies — that degrade TTR amyloid deposits and may be able to actually reverse disease.
Now that treatment of ATTR amyloidosis is becoming reality, recognizing the condition as early as possible has become critical. A review of advanced noninvasive tools for diagnosing cardiac amyloidosis appears in a recent Consult QD post.
ATTR amyloidosis can now be diagnosed without need for a cardiac biopsy, thanks to technetium-99m pyrophosphate scintigraphy. In a patient with suspected amyloidosis, if serum monoclonal light chain testing is negative (indicating that AL amyloidosis is unlikely) and the cardiac uptake of ATTR on scintigraphy is above a certain threshold, ATTR amyloidosis can be diagnosed with high sensitivity and specificity. At that point, genetic testing is needed to determine if ATTR is caused by an acquired wild-type variant or a hereditary mutant variant.
Advertisement
On the horizon is a plasma probe that can quantify misfolded transthyretin oligomers and help diagnose amyloidosis before symptoms develop. The probe is the focus of a research study to determine if it will be useful to regularly screen asymptomatic carriers of a hereditary ATTR mutation.
“This period reminds me of the time when effective HIV therapies first emerged,” says Dr. Hanna. “We believe the future will be combination therapy, such as treatment with an inhibitor of TTR production from the liver (such as patisiran or inotersen), a stabilizer of the TTR tetrameric protein (such as tafamidis) and possibly a TTR fibril disruptor to remove pre-existing amyloid deposits from the heart. The goal is to soon turn amyloidosis into a chronic disease that patients can live with for a long time.”
Dr. Hanna is Co-Director of Cleveland Clinic’s Amyloidosis Center, which coordinates involvement of specialists in cardiology, hematology/oncology, neurology, nephrology, gastroenterology and genetics for comprehensive patient care. The center also provides access to multiple investigator-initiated and multisite clinical trials.
Advertisement
Advertisement

Post hoc analysis of CLEAR Outcomes trial bolsters its case as a statin alternative

Large retrospective analysis may prompt prospective studies

How to talk about lifetime risk, treatment goals, Lp(a) testing, statin skepticism and more

A scannable recap of recent volumes and clinical metrics from Cleveland Clinic

Cleveland Clinic reports first U.S. series focused on use in this challenging setting

Large series confirms early and long-term survival advantages over partial pericardial resection

AVANT GUARD trial extends first-line role for ablation beyond paroxysmal atrial fibrillation

Maintain a high index of clinical suspicion and consider the underlying etiology