Locations:


Advanced COPD care benefits from diverse medical expertise and perspectives

Pediatricians and adolescent medicine specialists must play a role in curbing teen ENDS usage

Cleveland Clinic research emphasizes taking a holistic and individualized approach to care of septic shock

Pearls to reduce the strain of RSV, COVID-19 and influenza infections
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy

Management and diagnostic insights from an infectious disease specialist and a pulmonary specialist

Largest study examines factors affecting asthma exacerbations during and after pregnancy
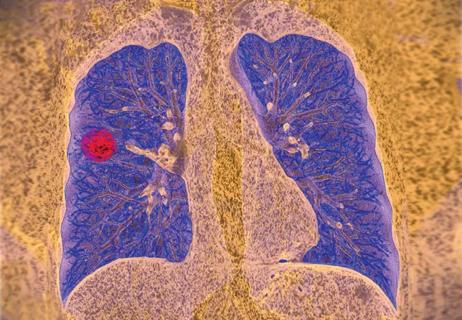
Resection, radiotherapy or ablation?

More than 30% of patients die when early dialysis is needed after surgery

Initial findings demonstrate improved symptoms and reduced steroid dependence

New developments offer providers more sophisticated options
Advertisement
Advertisement