Locations:

Pathophysiology, clinical features and disease management
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Patients who are born with the rare autosomal recessive disorder congenital erythropoietic porphyria (CEP) experience both cutaneous photosensitivity and hematologic abnormalities which can result in disfiguring and debilitating lesions, corneal scarring and transfusion-dependent anemia.
I recently published a review article on the pathophysiology, clinical presentation and management of CEP in Molecular Genetics and Metabolism.
CEP is caused by mutations in the uroporphyrinogen synthase (UROS) gene and, in three reported cases, by one specific mutation in the GATA1 gene, located on the X chromosome.
Mutations in these genes result in absent or markedly reduced uroporphyrinogen synthase (UROS) enzymatic activity, which leads to accumulation of non-physiologic and photoreactive porphyrinogens. Light exposure then elicits a phototoxic reaction, which damages erythrocytes and causes skin blistering.
In less than 10% of CEP patients, a disease-causing mutation cannot be attributed to the UROS or GATA1 genes, raising the question of whether additional gene(s) may have a role in CEP pathogenesis.
Patients with CEP have markedly deficient but present UROS activity. This leads to the accumulation of hydroxymethylbilane (HMB), the enzyme’s substrate, most of which is converted to the non-physiologic uroporphyrinogen I. Then uroporphyrin I builds up in patients’ bone marrow, erythrocytes, plasma and urine.
Patients’ skin and eyes become photosensitive. When exposed to sunlight and other sources of long-wave ultraviolet light, their skin blisters and easily tears.
Advertisement
Erythrocyte damage leads to hemolysis, which is almost always present. However, it is not always accompanied by anemia, because sometimes erythroid hyperplasia is sufficient to compensate for the increased rate of erythrocyte destruction.
The clinical presentation of CEP can be very variable and depends on each patient’s residual UROS enzyme activity. Usually, photosensitivity develops in early infancy. One of CEP’s earliest signs is the presence of reddish urine in the diaper which fluoresces under a Woods lamp.
Light exposure causes skin blisters which frequently rupture and often become infected. This process can lead to severe scarring, contractures and deformities such as loss of fingers and facial structures including eyelids, nose and ears. Increased hair growth is often present in face and on extremities. Patients often experience significantly diminished quality of life, functional status and behavioral health due to these long-term damages.
Mild to severe hemolysis leading to anemia is another common symptom of CEP. Increased uptake of abnormal erythrocytes causes secondary splenomegaly, which may contribute to the anemia and also may cause thrombocytopenia and leukopenia. Thrombocytopenia is sometimes associated with significant bleeding, and patients may benefit from splenectomy. Anemia can be severe, and some patients are transfusion-dependent.
Hematological complications and increased risk of infection can reduce overall life expectancy in patients with severe disease.
Advertisement
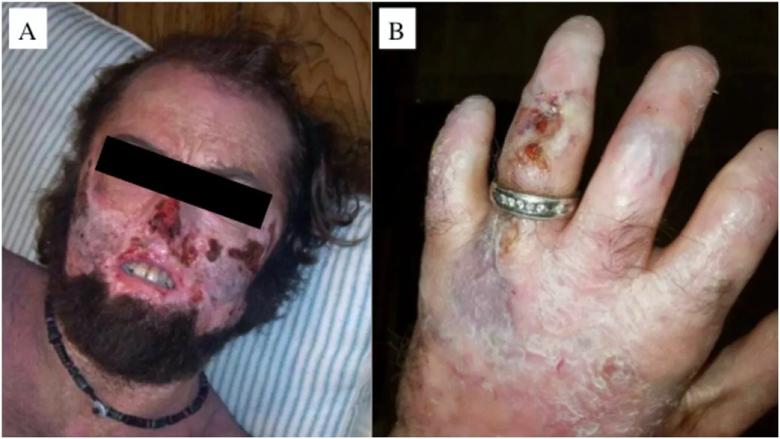
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/6c5d2387-bf2f-47c9-9b8e-0ec6a6da7131/erwin-face_jpg)
Male 34 year-old CEP patient with severe cutaneous manifestations of face (left) and hand (right). Note the extensive skin erosions in the face, destruction of the nasal cartilage, sclerodermoid changes of the mouth, and erythrodontia. The patient also has significant shortening of the digits due to photomutilation and contractures of the joints, along with erosions and scleroderma-like thickening of the skin.
Physicians should consider CEP in the fetus or infant if there has been a diagnosis of non-immune hydrops fetalis, in which the amniotic fluid around the fetus will be pink, dark-red or brown, and should be examined for porphyrins. Infants with pink to dark-red urine-stained diapers should also be evaluated for CEP.
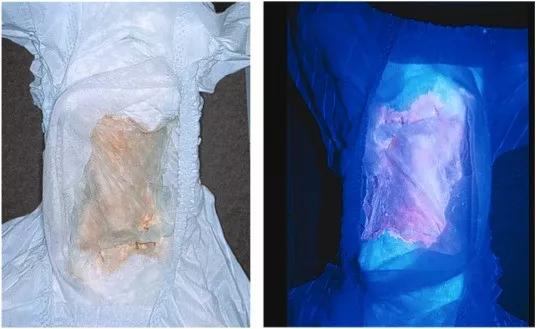
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/87559642-f303-44d9-a0a7-b607b31ee4a8/erwin-diaper_jpg)
Diaper of an infant with CEP. The patient’s urine has stained the diaper a reddish color due to the accumulated uroporphyrin I and coproporphyrin I (left). The accumulated porphyrins fluoresce when illuminated with ultraviolet light (right).
Clinicians should also consider CEP in children or adults with red urine or skin blistering following sunlight or other long-wave ultraviolet light exposure. Some less severe cases present in adult life with skin lesions and/or mild anemia.
To establish the biochemical diagnosis of CEP, one should observe significantly elevated levels of uroporphyrin I and coproporphyrin I in urine, erythrocytes or amniotic fluid in addition to high fecal concentrations of coproporphyrin I. Sequencing of the UROS gene is recommended to confirm the diagnosis. This also helps to predict expected disease severity and thus guide management decisions.
Advertisement
Bone marrow or hematopoietic stem cell transplantation remains the only curative treatment approaches for CEP. Both are associated with significant morbidity and mortality and need to be considered carefully. For patients who do not undergo transplant, avoidance of sun and light exposure is the most important tool of disease management.
Frequent blood transfusions may be warranted for patients with severe hemolysis. Transfusions every two to four weeks can suppress erythropoiesis and decrease the production of porphyrin, thus reducing photosensitivity. If the patient’s hematocrit stays above 35%, and they can take chelators for the iron overload, such therapy is likely to be helpful.
Various treatment approaches are under investigation, including genetically modified hematopoietic stem cells and proteasome inhibitors. However, much more work is necessary to offer more and better therapeutic options to patients.
Dr. Erwin is staff in the Genomic Medicine Institute.
Images republished with permission from Elsevier.
Advertisement
Advertisement

GenT framework aims to improve drug development with focus on entire genes, not individual mutations

Study identifies Ketorolac as a potential repurposable drug

The relationship between MTHFR variants and thrombosis risk is a complex issue, but current evidence points to no association between the most common variants and an elevated risk

One-time infusion of adenovirus-based therapy is designed to restore heart muscle function
Cleveland Clinic researchers receive $2 million grant from the National Institutes of Health

New Cleveland Clinic fellowship fosters expertise in the genetics of epilepsy
Integrates genetic and clinical data to distinguish from GEFS+ and milder epilepsies

First-of-kind study aims to enable prevention of brain disorders before symptoms appear