Locations:

A review of vascular calcification, soft tissue calcification and calciphylaxis
Editor’s note: This is part one of a two-part series on extraosseous calcification in kidney disease. The original article, including a full list of references, was published in the Cleveland Clinic Journal of Medicine and can be accessed here.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
By Korey Bartolomeo, DO; Xin Yee Tan, MD; and Richard Fatica, MD
Chronic kidney disease, defined as an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2 or structural kidney damage sustained over 3 months, is increasing in prevalence worldwide. It is estimated to affect between 2% and 17% of all adults, and the United States is at the high end of this prevalence range.1
As chronic kidney disease progresses, it leads to higher rates of bone mineral disease, a systemic disorder involving the following:
Patients with end-stage kidney disease are at high risk of complications from disorders of bone metabolism, which are strongly associated with increased rates of cardiovascular and all-cause mortality.3–6
Extraosseous tissue calcification can involve both vascular tissues (arteries and heart valves) and soft tissues. A variety of terms have been used to describe it, based on the location and the type of tissue involved (Table 1), but subclassifying it precisely and studying its prevalence are challenging because its presentation is heterogeneous.

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/93d6f41e-53eb-4f30-b100-a4bdc87585d2/Table-1-inset_jpg)
Serum calcium and phosphate levels are kept under tight control by regulatory hormones released by various organs, with complex feedback mechanisms (Figure 1).
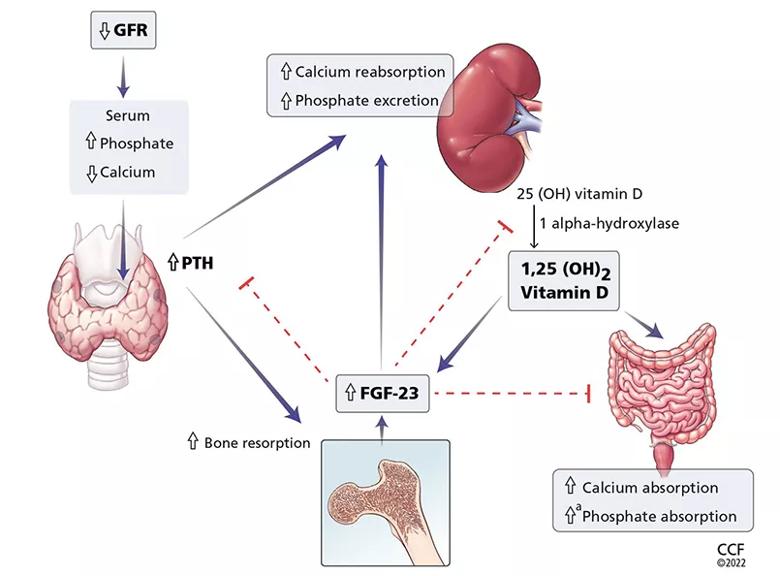
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/42f2c156-5a2c-44f9-a3bc-b86a3fa986ee/F1_large-inset_jpg)
Figure 1. Calcium and phosphate metabolism in chronic kidney disease. Decreased glomerular filtration rate (GFR) leads to changes in serum calcium and phosphate, triggering release of parathyroid hormone (PTH) from the parathyroid glands and fibroblast growth factor 23 (FGF-23) from osteoblasts and osteocytes. These hormones have complex downstream effects on the kidney, gut, and bone, both from direct effects on the tissue and from indirect effects through modulation of enzyme activity in vitamin D conversion.
Advertisement
aMinimally increased.
25(OH) vitamin D = 25-hydroxycholecalciferol; 1,25(OH)2 vitamin D = 1,25-dihydroxycholecalciferol
Interestingly, both calcium and phosphate are regulated by the same hormone, i.e., PTH.7,8 When serum calcium levels are low and serum phosphate levels are high, the parathyroid glands release more PTH, which acts in several organs to raise the calcium and, on the whole, to lower the phosphate levels.
In the kidney, PTH directly increases calcium reabsorption in the distal tubule and loop of Henle and increases phosphate excretion by inhibiting its reabsorption in the proximal tubule.9,10 Also in the kidney, PTH upregulates production of 1 alphahydroxylase, leading to increased conversion of active vitamin D (1,25-dihydroxycholecalciferol) from its precursor, 25-hydroxycholecalciferol. In turn, in the intestine, active vitamin D increases the absorption of calcium and to a lesser degree phosphate, and in the bone, it has direct actions on both osteoblasts and osteocytes, promoting maturation, expression of skeletal hormones such as fibroblast growth factor 23 (FGF-23), and proper mineralization.11,12
FGF-23 is an important skeletal hormone that lowers phosphate levels by promoting its wasting (i.e., suppressing its reabsorption) in the kidney, suppressing its absorption in the intestine, and, in a negative feedback loop, lowering both PTH and 1,25-dihydroxycholecalciferol production.13 Klotho, a protein that has multiple effects in many tissues, facilitates binding of FGF-23 to FGF receptor 1 in the kidney, leading to fewer phosphate receptors in the proximal convoluted tubules, more phosphate excreted in the urine, and lower serum phosphate levels.14 The net effect of these interactions is homeostatic balance in serum calcium and phosphate levels.
Advertisement
In chronic kidney disease, nephrons are progressively lost. Among the ill effects is a higher phosphate level, which in turn upregulates production of FGF-23 by the osteocytes and osteoblasts and leads to bone mineral disease (Figure 1). Bone mineral disease can begin early in the course of chronic kidney disease,15 when the eGFR may still be as high as 69 mL/min/1.73 m2. Meanwhile, klotho production is downregulated, so that less FGF-23 binds to its receptor in the kidney,16,17 less 1 alpha-hydroxylase and active vitamin D are produced, and more phosphate is reabsorbed in the proximal convoluted tubule.18,19
As chronic kidney disease progresses to its end stage, FGF-23 levels keep getting higher, and the elevation is accompanied by other calcium-phosphate axis derangements such as excess PTH release, decreased 1,25-dihydroxycholecalciferol, and increased sclerostin (an inhibitor of bone formation).20,21 Together, these derangements lead to the clinical manifestations described below.
Vascular calcification
Vascular calcification is an active process involving de-differentiation of vascular smooth muscle cells. It begins with amorphous development of calcium phosphate nanocrystals in conjunction with other calcium-regulatory proteins in the wall of the artery.22 Deposition of these nanocrystals can begin in the intima of the artery near sites of cholesterol buildup, either progressing into the media or beginning in the media itself, the latter of which is most specific to kidney disease.
Advertisement
In end-stage kidney disease, progression of vascular calcification occurs earlier than in normal aging and is likely driven by hyperphosphatemia, a positive calcium balance, inflammation, and dysregulation between pro-calcification and anticalcification regulatory factors. An in-depth discussion of the pathogenesis of vascular calcification is beyond the scope of this paper and can be found elsewhere.23
Soft tissue calcification
Soft tissue calcification is fairly common in chronic and end-stage kidney disease, but only a small number of patients develop tumoral calcinosis, characterized by massive calcium phosphate deposition in periarticular locations predisposed to microtrauma.
Tumoral calcinosis is well described in families, with autosomal-recessive inheritance stemming from a number of genes, including loss-of-function mutation of FGF23 and missense mutation of alpha-Klotho, contributing to the hyperphosphatemia.24 Hyperphosphatemia is likely a necessary contributor to these familial forms of tumoral calcinosis, but it may also explain their presence in chronic and end-stage kidney disease, stemming from local tissue production or from exogenous phosphate retention.25,26
Calciphylaxis is intensely painful, unlike other presentations (Figure 2).27 It is most commonly seen in adipose-dense tissues but can develop centrally and in appendicular areas, including the genital regions. Skin lesions can vary from induration to ulceration with eschar formation.28 Its diagnosis is predominantly clinical. A skin biopsy to the depth of the subcutaneous tissue can aid diagnosis but poses significant procedural risks that include pain intensification, poor healing, and secondary infection.29
Advertisement
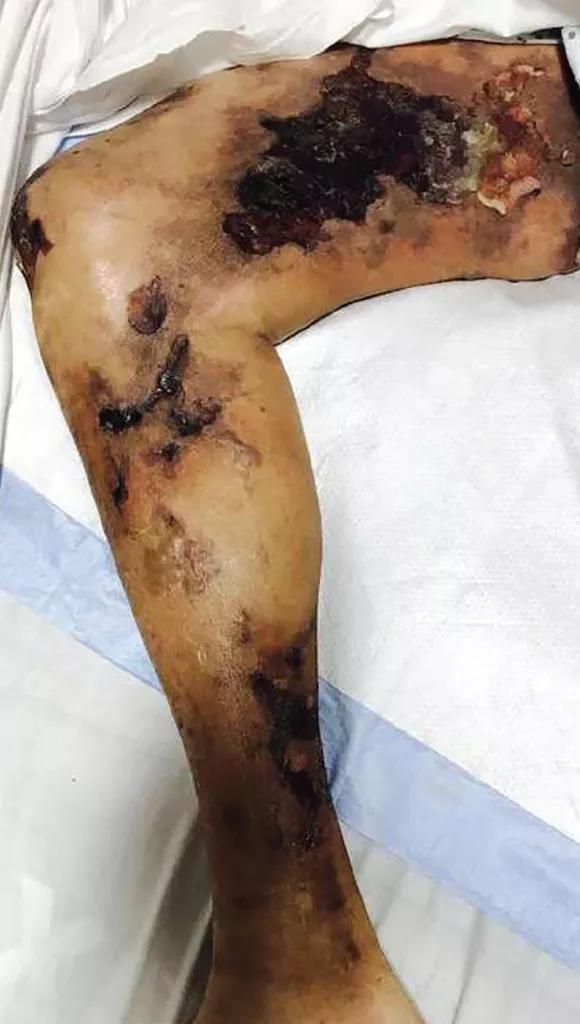
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/7a42d71a-e507-49e1-a04d-1b2baf1eef8b/F2_large-inset-1-580x1024_jpg)
Figure 2. Calciphylaxis in a 51-year-old man with end-stage kidney disease. From reference 27.
Soft tissue calcifications, in contrast, are usually painless, unless radicular symptoms develop from mass effect. Instead, there is typically a decrease in range of motion of the affected joints,30 of which (in descending order of frequency) the hip, elbow, shoulder, foot, and wrist are most commonly affected (Figure 3).31 Soft tissue calcifications tend to be formally diagnosed based on the location of the calcium deposition, in addition to morphologic descriptions to rule out cancer mimickers.
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/662ccdb0-4917-49a0-9d98-1361fa0c3278/F3_large-inset-541x1024_jpg)
Figure 3. Radiography shows calcified masses (arrows) in a 47-year-old woman with tumoral calcinosis. From reference 30.
Vascular calcification. Traditional risk factors that predict atherosclerotic calcification do not fully explain the high prevalence of vascular calcification in patients with chronic and end-stage kidney disease. Additional potentially modifiable risk factors related to kidney disease or its treatment have been shown to accelerate calcification (Table 2).32

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/15a574b0-7144-48e2-b1d2-71062408305b/Table-2-inset_jpg)
Table 2. Risk factors associated with vascular calcification
Read on for part two, which includes strategies for management, including a discussion about randomized trials over the last 20 years that aim to settle the debate on calcium-based vs non–calcium-based phosphate binders and cardiovascular disease.
Disclosures
Dr. Fatica has disclosed working as an advisor or review panel participant for Natera Inc and REATA Pharmaceuticals. The other authors report no relevant financial relationships which, in the context of their contributions, could be perceived as a potential conflict of interest.
References
Advertisement

Pediatric urologists lead quality improvement initiative, author systemwide guideline

Fixed-dose single-pill combinations and future therapies

Reproductive urologists publish a contemporary review to guide practice

Two recent cases show favorable pain and cosmesis outcomes

Meta-analysis assesses outcomes in adolescent age vs. mid-adulthood

Proteinuria reduction remains the most important treatment target.

IgA nephropathy is a relatively common autoimmune glomerular disease that can be diagnosed only by biopsy

Oncologic and functional outcomes are promising, but selection is key