Locations:

Four genomic clusters identified
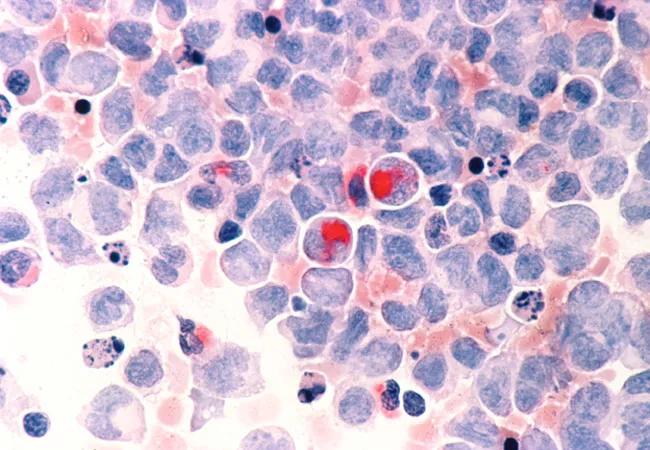
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/cf935ab0-4624-407b-9b35-d03e132e2a7c/nci-vol-2336_650x450_jpg)
nci-vol-2336_650x450
Cleveland Clinic Cancer Center investigators successfully applied machine learning to improve the subclassification and prognostication in acute myeloid leukemia (AML), in a study led by hematologic oncologist Jaroslaw Maciejewski, MD, PhD. Study findings were presented at the 2020 Annual Meeting of the American Society of Hematology (ASH) and garnered significant attention from meeting attendees for their innovative application of machine learning technology in AML.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Below, Hassan Awada, MD, study lead author and research fellow in the Department of Translational Hematology and Oncology Research, explains the relevance of these latest findings and their impact on the field moving forward.
A: Our research was inspired by the lack of a deeper understanding of the complex pathogenetic links and the overlapping mutational spectra of pathomorphological AML subtypes including de novo/primary AML (pAML) and secondary AML (sAML) evolving from antecedent myeloid neoplasm. Therefore, we aimed to explore the use of distinct genomic markers, uncovered by machine learning techniques, to objectively sub-classify AML patients, irrespective of the availability of clinicopathological information.
A: Initially, we performed a supervised analytical approach using logistic regression analyses that revealed significant molecular associations defining pAML vs sAML. However, when these markers were used to reassign pathomorphological AML subtypes, the approach yielded only 74% accuracy.
Subsequently, we explored other machine learning methods including Bayesian latent class analysis (BLCA). The unsupervised clustering of pAML and sAML cases using BLCA applied to unbiased genomic data only (cytogenetics and gene mutations) uncovered four novel genomic AML clusters of distinct prognoses. We then generated a random forest model to extract the distinct genomic features defining each AML cluster. The resulting multiclass classifier yielded across-validation accuracy of 0.97, which is equivalent to 97% performance accuracy when using these genomic features to reassign the four genomic AML clusters.
Advertisement
A: AML diagnostics has been traditionally based on phenotype and morphological assessment, but more recently, cytogenetics and molecular genetics have been integrated to better define AML subtypes, assess prognostication, guide treatment decisions and drive the future discovery of targeted therapeutics based on disease genotype. However, the combinatorial complexity and heterogeneity of AML genetics have, so far, precluded additional progress in this field. Machine learning techniques can essentially optimize our efforts of decoding high dimensional genomic interactions in AML, and thus augment human intelligence to accurately define AML genomics, objective subtypes and risk groups.
A: I strongly believe that advanced machine learning techniques will lead future research efforts aimed at discovering complex phenotype-genotype correlations and predicting population-level outcomes not just in AML, but in other fields of medical research.
A: Cluster-1/low risk AML group was defined by a 100% presence of NPM1 mutations with frequently co-mutated DNMT3A, FLT3ITD, IDH2R140Q and high prevalence of normal cytogenetics. Cluster-2/intermediate-low AML group had the highest frequency of CEBPAbiallelic, IDH2R172K, FLT3ITD and FLT3TKD mutations occurring in the absence of NPM1 mutations. Cluster-3/intermediate-high AML group had the highest frequency of mutations in ASXL1, BCOR/L1, DNMT3A without NPM1, EZH2, RUNX1 and splicing factor gene mutations in SF3B1, SRSF2 and U2AF1. Finally, Cluster-4/high risk AML group had the highest prevalence of abnormal cytogenetics, mainly complex karyotype including -5/del(5q), -7/del(7q), -17/del(17p) and the highest rate of TP53 mutations (70% of cases). Our results were internally and externally validated using an independent cohort.
Advertisement
A: This study is the first to apply innovative machine learning methods to such a large cohort of AML patients. It also identified four novel AML genomic clusters of discrete prognoses, irrespective of any clinicomorphological data. Hence, this work proposes the reevaluation of traditional pathomorphological classification of pAML vs sAML due to their overlapping mutational spectra and shared pathogenesis and supports the shift towards a molecularly informed AML subtyping that is more reflective of disease pathogenesis.
Image: Human cells with acute myelocytic leukemia (AML) in the pericardial fluid, shown with an esterase stain at 400x. Source: NCI Visuals Online.
Advertisement
Advertisement

First full characterization of kidney microbiome unlocks potential to prevent kidney stones

Researchers identify potential path to retaining chemo sensitivity
Large-scale joint study links elevated TMAO blood levels and chronic kidney disease risk over time
Investigators are developing a deep learning model to predict health outcomes in ICUs.

Preclinical work promises large-scale data with minimal bias to inform development of clinical tests

Cleveland Clinic researchers pursue answers on basic science and clinical fronts

Study suggests sex-specific pathways show potential for sex-specific therapeutic approaches

Cleveland Clinic launches Quantum Innovation Catalyzer Program to help start-up companies access advanced research technology