Locations:

Novel drug could greatly improve patient outcomes
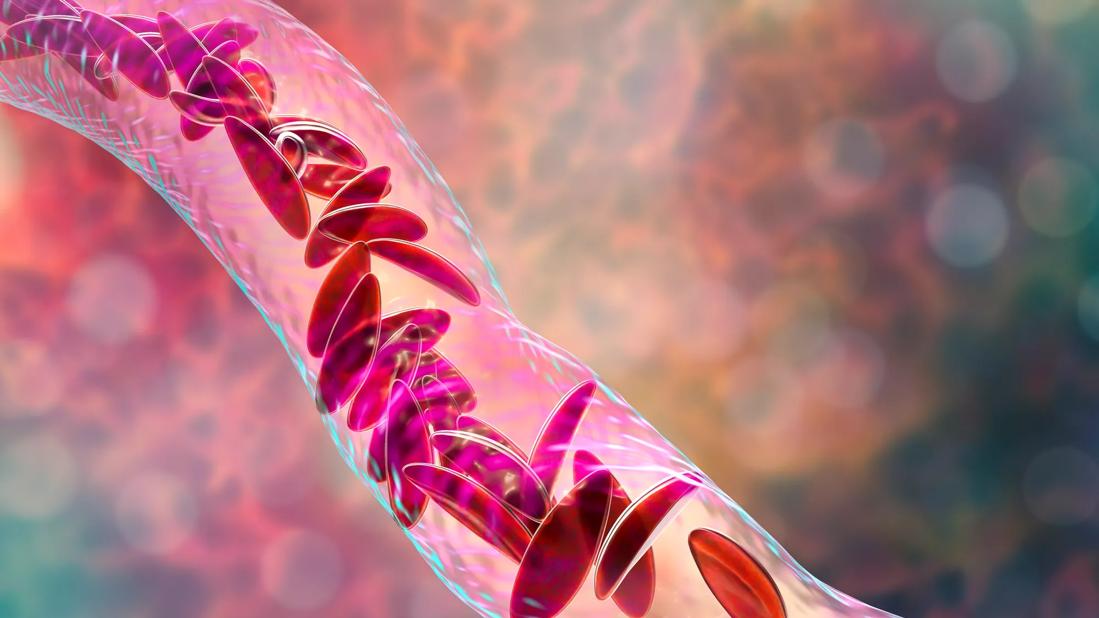
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/5672314f-8ce6-4f9d-afe4-5edc45b8dce7/GettyImages-1059145434-jpg)
SCD
A phase 1 clinical trial has demonstrated safety and preliminary evidence of efficacy for a new combination oral drug to treat sickle cell disease (SCD).
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Affecting 4.4 million people worldwide, SCD is a congenital hemolytic anemia caused by a defect in the gene that directs the production of the beta globin part of hemoglobin. The mutated sickle hemoglobin (HbS) polymerizes and precipitates in red blood cells (RBCs), destroying the cells and blocking blood vessels, and causing multiorgan damage, severe pain and compromised immunity.
The prognosis for SCD patients is poor: the average life expectancy in developed countries is 40 to 60 years and, in less developed countries, most children with SCD do not make it to adulthood. “Sickle cell disease is a terrible illness that causes great suffering worldwide,” says Yogen Saunthararajah, MD, staff in the Department of Hematologic Oncology and Blood Disorders at Cleveland Clinic Cancer Center.
There are only two FDA-approved medications to treat SCD, hydroxyurea and glutamine, and both have limited impact. One focus of research efforts has been to develop drugs that increase expression of fetal hemoglobin (HbF). HbF is usually expressed in RBCs until infancy and intercalates with HbS to prevent its polymerization and precipitation. By interfering with HbS polymerization, HbF is the most powerful known modulator of this root cause of SCD pathophysiology. People with naturally high levels of HbF beyond infancy have some protection from SCD complications and nearly normal life spans.
One way of inducing HbF is bone marrow stress; HbF is enriched during the recovery from stress. Cytotoxic drugs can create this stress, which led to the approval of the oral cytotoxic drug hydroxyurea for use in SCD. Unfortunately, this cytotoxic mechanism of action is not sustainable, since it destroys bone marrow precursors needed to make red blood cells over the long term.
Advertisement
“Hydroxyurea produces temporary changes; we badly need more durable, effective treatments,” says Dr. Saunthararajah. Glutamine, the other FDA-approved medication, is essentially a nutritional supplement and does not address root-cause pathophysiology.
The shutting down of HbF from infancy onward is mediated by the enzyme DNA methyltransferase 1 (DNMT1). The drug decitabine, which is used to treat myelodysplastic syndrome, can deplete DNMT1. Unfortunately, decitabine is rapidly inactivated in the body and is not meaningfully absorbed by the oral route. Dr. Saunthararajah and his collaborators developed a novel combination of decitabine and another drug, tetrahydrouridine, which enables its absorption and distribution through the body after oral ingestion.
Dr. Saunthararajah and colleagues at the University of Illinois conducted the first-in-human clinical trial of this new oral drug in 25 SCD patients with severe, symptomatic SCD that had not improved with standard treatment with hydroxyurea. They were divided into five dose-levels, with three patients in each group receiving the medication regimen and two the placebo for eight weeks.
Over the course of the trial, a therapeutic dose of oral THU-decitabine was established that was safe, well-tolerated and which substantially increased fetal and total hemoglobin as well as improved biomarkers of hemolysis, coagulation and inflammation. The findings are reported in PLOS Medicine.
“We believe that the results are very significant. This drug combination offers a mechanistically rational, potentially lasting way to change the natural history of sickle cell disease by addressing its root-cause pathophysiology,” says Dr. Saunthararajah.
Advertisement
The next step is a phase 2-3 clinical trial scheduled to begin in 2018.
Photo Credit: ©Russell Lee
Advertisement
Advertisement

Patients on ibrutinib were more than twice as likely to experience atrial fibrillation or heart failure than those on second-generation alternatives.

Expert recommendations introduce use of targeted agents

Combination therapy may soon represent new standard of care

Research findings offer clues for improving disease outcomes in men

Creating a safe space for patients

Long-term immune effects reshape preventative strategies and timelines

Large-scale database also reveals potential for immunotherapy to protect against cancer

Findings may help guide discussions around prognosis and allogeneic stem cell transplantation