Locations:

Insights for diagnosing, assessing and treating
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/3b07f596-a2ba-4134-ab24-528faaa160cb/24-PUL-4507382-CQD-Portopulmonary-Hypertension-Hero-967x544-1_jpg)
24-PUL-4507382-CQD-Portopulmonary-Hypertension-Hero-967×544
Written by Mamta S. Chhabria, MD, Leela Krishna Teja Boppana, MD, Gaurav Manek, MD and Adriano R. Tonelli, MD, MSc
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Note: This article is reprinted from the Cleveland Clinic Journal of Medicine (2023;90[10]:632-639)
For reasons that are not entirely clear, some patients who have portal hypertension go on to also have pulmonary arterial hypertension, a grim but fortunately rare combination called portopulmonary hypertension.
Portopulmonary hypertension is important to recognize, both because it has a poor prognosis and because it can affect a patient’s eligibility for liver transplant.
This article reviews the key aspects of screening, diagnosis and treatment of patients with portopulmonary hypertension and highlights the various pulmonary hemodynamic patterns encountered in patients with liver disease.
Portal hypertension is characterized by a high-pressure gradient (> 5 mm Hg) between the portal venous system and the hepatic veins.2
It is usually caused by liver cirrhosis, although noncirrhotic causes such as congenital hepatic fibrosis, sarcoidosis and schistosomiasis are occasionally seen. It can be recognized clinically by its classic signs and sequelae such as gastroesophageal varices, portal hypertensive intestinal vasculopathies, ascites, spontaneous bacterial peritonitis, hepatic hydrothorax and hepatorenal syndrome.
Pulmonary arterial hypertension has a specific hemodynamic profile called precapillary pulmonary hypertension, defined by the following:
Advertisement
The exact prevalence of portopulmonary hypertension in the United States is difficult to determine, but it is not common.6-8 McDonnell et al9 reported that patients with hepatic cirrhosis had a prevalence of pulmonary arterial hypertension of 0.73%. In the United States and Europe, the prevalence of pulmonary arterial hypertension ranges from 15 to 50 per million, with portopulmonary hypertension accounting for 5% to 15% of cases.10 In a prospective study of 1,235 patients undergoing liver transplant in the United States, approximately 5% met the criteria for portopulmonary hypertension.11
The incidence of portopulmonary hypertension will likely increase as our population ages and as the prevalence of cirrhosis increases. In North America, the prevalence of cirrhosis has increased 1.5-fold to 2-fold over the past two decades.12
The pathophysiology of portopulmonary hypertension remains unclear but may involve several factors, including the following:
Genetic predisposition. Several genetic variants are thought to play a role, including single-nucleotide polymorphisms in the genes coding for estrogen receptor 1, aromatase, phosphodiesterase 5, angiopoietin 1 and calcium-binding protein A4.13,14
Hyperdynamic circulation. Patients with chronic liver disease and cirrhosis have high cardiac output and low systemic vascular resistance. This hyperdynamic circulatory state may contribute to higher pulmonary vascular shear stress (frictional force of blood flow on the endothelium),15 which may injure endothelial cells and activate genes that participate in vascular remodeling.
Advertisement
Inflammation. Bacteria can enter the portal circulation through disruptions in the intestinal barrier. Bacterial lipopolysaccharides can activate Toll-like receptors on immune cells, causing them to release inflammatory cytokines such as interferon gamma and interleukin 6, which have been implicated in the pathogenesis of pulmonary arterial hypertension.15,16
Imbalance of vasoconstrictive and vasodilatory mediators. Portosystemic shunts develop as a result of portal hypertension.17 These shunts may allow vasoactive substances in the blood to evade hepatic metabolism and enter the pulmonary circulation, causing vasoconstriction and endothelial remodeling.15 In addition, levels of specific mediators such as bone morphogenetic proteins 9 and 10, which are responsible for maintaining vascular quiescence, have been found to be lower in patients with portopulmonary hypertension than in healthy controls.18,19
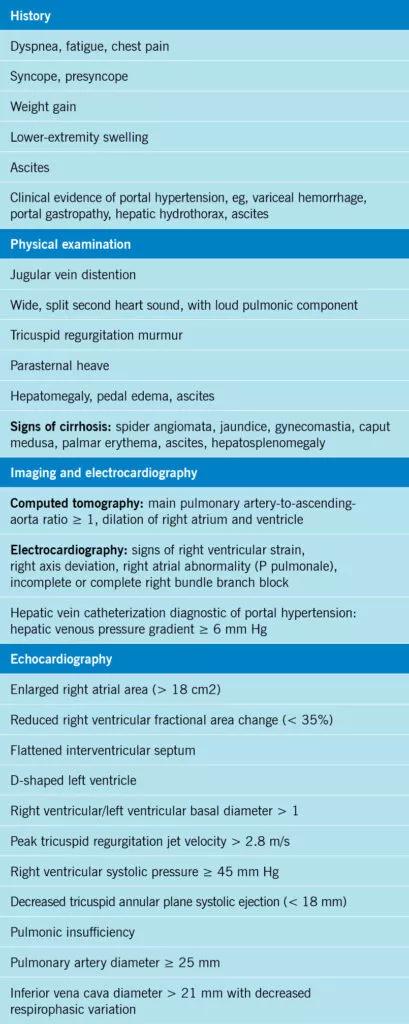
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/ba1cfee1-0499-47d5-99a0-ecae1f1fd58b/24-PUL-4507382-CQD-Portopulmonary-Hypertension-Chart-1-409x1024_jpg)
Table 1
In general, portal hypertension precedes the development of portopulmonary hypertension by several years.20 Suspect portopulmonary hypertension in patients with portal hypertension and chronic liver disease who have dyspnea on exertion, chest pain or exertional syncope or near-syncope (Table 1). Patients may also present with signs suggesting right heart failure such as jugular venous distention, edema, ascites, a second heart sound that is wide and split, and a murmur of tricuspid regurgitation.21
Patients with portal hypertension, particularly those being evaluated for liver transplant, should be screened for portopulmonary hypertension with transthoracic echocardiography.5 The right ventricular systolic pressure as estimated by echocardiography can differ widely from that measured directly by right heart catheterization.11 Patients awaiting liver transplant should be screened for portopulmonary hypertension with echocardiography at least annually, although the optimal interval is unknown.5
Advertisement
In patients with liver disease, three hemodynamic abnormalities can exist alone or in combination, and some patients have all three (Table 2)1:
Right heart catheterization is essential for identifying the predominant hemodynamic pattern.
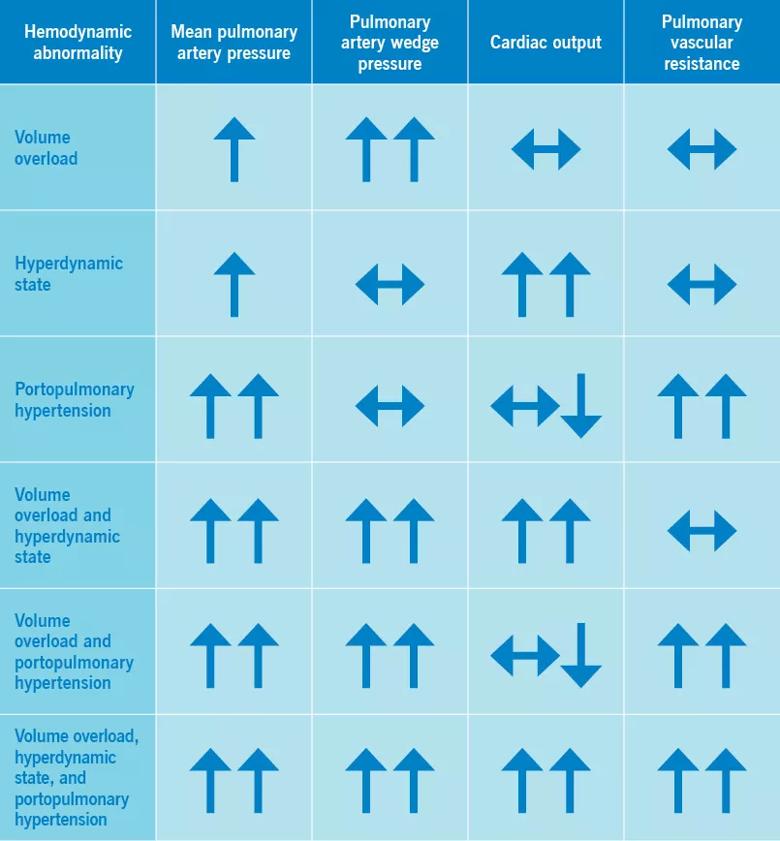
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/8b94fb90-e343-402f-8dbe-fc630452f0fd/24-PUL-4507382-CQD-Portopulmonary-Hypertension-Chart-2_jpg)
Table 2
Although a hyperdynamic state is inherent in liver disease, it can also be caused or exacerbated by conditions such as anemia, obesity, thiamine deficiency, systemic arteriovenous shunts and hyperthyroidism.22 Similarly, a volume overload state can be present in patients with concomitant renal disease or left heart failure (systolic or diastolic, or both). And precapillary pulmonary hypertension, suggestive of portopulmonary hypertension, can also be seen in patients with scleroderma, congenital heart disease, drug or toxin exposure, lung diseases, hypoxia, chronic thromboembolic disease and sarcoidosis.5
Patients with hyperdynamic circulation and volume overload can undergo liver transplant without pulmonary hypertension therapies. However, liver transplant is contraindicated in patients with portopulmonary hypertension who have persistently elevated pulmonary vascular resistance despite pulmonary hypertension treatment. In 2021, the Organ Procurement and Transplantation Network modified its criteria and now allows liver transplants for patients with portopulmonary hypertension with either of the following two hemodynamic patterns after treatment:
Advertisement
Although the 2019 World Symposium on Pulmonary Hypertension decreased the mean pulmonary arterial pressure threshold for the diagnosis of pulmonary hypertension from 25 mm Hg or higher to higher than 20 mm Hg,4 and the 2023 guidelines lowered the pulmonary vascular resistance threshold from three or more WU to more than 2 WU,5 inclusion criteria in studies of portopulmonary hypertension were based on the older definitions.24 The current hemodynamic criteria for portopulmonary hypertension remain the following:
Important goals of therapy include symptom relief and improvements in functional capacity and quality of life. General management includes supplemental oxygen for hypoxemia (resting, exercise-induced, or nocturnal), diuretics for fluid overload and an exercise program. Specific treatment includes pulmonary vasodilator therapy and, ideally, liver transplant once the pulmonary hemodynamic profile is optimized.25
Medications specifically for pulmonary arterial hypertension help reduce pulmonary vascular resistance while improving right ventricular function.
Importantly, these medications reduce pulmonary vascular resistance more than they decrease the mean pulmonary artery pressure because they also increase cardiac output, which can partially offset the expected improvement in mean pulmonary artery pressure.26 In the Portopulmonary Hypertension Treatment With Macitentan—a Randomized Clinical Trial (PORTICO),27 patients treated with macitentan had a decrease in pulmonary vascular resistance of 37% at 12 weeks, while the mean pulmonary artery pressure dropped 14% and the cardiac index increased 19%.
A unique role of pulmonary arterial hypertension therapies in patients with portopulmonary hypertension is to facilitate liver transplant. A meta-analysis of 26 observational and case-controlled studies in 1,019 patients showed that pulmonary hypertension therapies in patients with portopulmonary hypertension improved their pulmonary hemodynamic numbers, and more importantly, 44% became eligible for liver transplant.28
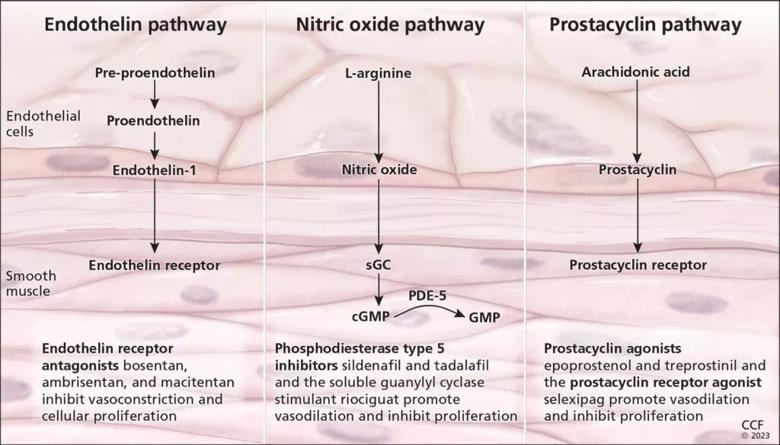
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/44e5dd9c-1058-4b5e-8909-85e609874768/24-PUL-4507382-Figure-1-1024x584_jpg)
Figure 1
Current drugs for pulmonary arterial hypertension belong to several classes (Figure 1)25,29:
All the above PAH-specific drugs are metabolized in the liver, except for epoprostenol, which is rapidly hydrolyzed in blood. Individual medications in these classes have different dosing requirements in patients with cirrhosis. A detailed description of their use in the context of liver cirrhosis was previously published by our group.25
Calcium channel blockers are generally not used in patients with portopulmonary hypertension because they can worsen hypotension and exacerbate portal hypertension.30

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/4280c0da-63e6-4636-b644-a8c972cb0b91/24-PUL-4507382-CQD-Portopulmonary-Hypertension-Chart-3-430x1024_jpg)
Table 3
In case reports and small case series, patients with portopulmonary hypertension showed improvements in their pulmonary hemodynamics with pulmonary hypertension therapies. A prospective cohort study examined 637 patients with portopulmonary hypertension, of whom 90% were treated with pulmonary arterial hypertension-specific therapies (74% received monotherapy), resulting in significant improvement in functional class and hemodynamic parameters. Notably, 63 patients underwent liver transplant, of whom 60 (95%) were on pulmonary hypertension therapies as a bridge to transplant.31 Furthermore, a retrospective study of 21 patients with portopulmonary hypertension showed that early initiation of parenteral epoprostenol therapy allowed 52% of them to become eligible for liver transplant within one year.32
Unfortunately, the side effects of pulmonary arterial hypertension-specific therapies often overlap with signs and symptoms of liver disease such as nausea, vomiting, anorexia, and edema, limiting the aggressiveness of this treatment.25
Only a few studies have tested the impact of pulmonary arterial hypertension therapies in patients with portopulmonary hypertension (Table 3).27,33-36 At the time of this writing, only one randomized controlled trial in portopulmonary hypertension (PORTICO)27 has compared a pulmonary arterial hypertension therapy (macitentan) and placebo. The Pulmonary Arterial Hypertension Soluble Guanylate Cyclase–Stimulator Trial 133 randomized patients with pulmonary arterial hypertension to riociguat vs placebo and included a subgroup of patients with portopulmonary hypertension.33-35 In addition, there is an open-label observational trial in portopulmonary hypertension using ambrisentan.36 In general, patients with portopulmonary hypertension are excluded from trials in pulmonary arterial hypertension owing to hepatic safety concerns and unpredictable blood levels of medications in the context of chronic liver failure.
Patients with suspected or known portopulmonary hypertension should be referred to a pulmonary hypertension center of excellence with multidisciplinary care, as their care is complex. The medications are poorly tolerated and need frequent changes in type and dosage, and patients need serial evaluations and treatment optimizations to achieve or maintain eligibility for liver transplant.
The five-year mortality rate exceeds 60% even with treatment,37 and many patients die of complications of their liver disease.38 In the Registry to Evaluate Early and Long-term Pulmonary Arterial Hypertension Disease Management, patients with portopulmonary hypertension had lower survival rates than those with idiopathic or familial pulmonary arterial hypertension (67% vs 85% at two years, and 40% vs 64% at five years).39
In a multivariable model of portopulmonary hypertension in patients from our institution, the Model for End-stage Liver Disease-Na score, resting heart rate and hepatic encephalopathy were independent predictors of death, while the severity of portopulmonary hypertension did not predict pretransplant mortality risk.37 Similarly, other investigators showed that severity of cirrhosis negatively affected outcomes,40 and that the prognosis for patients with portopulmonary hypertension prognosis is poor if they do not receive a liver transplant, despite the use of therapies for pulmonary arterial hypertension.41 Patients with portopulmonary hypertension do better after liver transplant, with improvement in hemodynamics and decreased need for pulmonary vasodilators.42 Therefore, efforts should focus on facilitating liver transplant whenever possible.25,36
It is essential to differentiate portopulmonary hypertension from other types of pulmonary hypertension, as postcapillary pulmonary hypertension does not appear to have a negative impact on survival after liver transplant.43
REFERENCES
Advertisement

Overexposure to the common alloy can lead to an autoimmune response called sensitization

Research demonstrates cirrhosis regression in one-third of patients, with higher rates using alternative assessment

Rapid growth and collaboration have advanced the Lung Nodule Program, but patient engagement barriers persist

Blood pressure decreases equally in study groups receiving monthly or one-time doses

A look into recent developments in the diagnosis of LAM and its clinical presentation

A review of treatment options for patients who may not qualify for surgery

Insights on ex vivo lung perfusion, dual-organ transplant, cardiac comorbidities and more

Multidisciplinary framework ensures safe weight loss, prevents sarcopenia and enhances adherence