Locations:


The case for continued vigilance, counseling and antivirals

Study sheds light on how clinicians addressed their patients’ pain and insomnia during the pandemic
Patients report improved sense of smell and taste
Clinicians who are accustomed to uncertainty can do well by patients
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy

Unique skin changes can occur after infection or vaccine

Cleveland Clinic analysis suggests that obtaining care for the virus might reveal a previously undiagnosed condition

As the pandemic evolves, rheumatologists must continue to be mindful of most vulnerable patients
Early results suggest positive outcomes from COVID-19 PrEP treatment
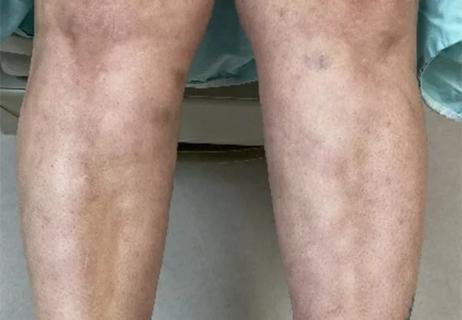
Could the virus have caused the condition or triggered previously undiagnosed disease?

A Q&A with infectious disease expert Eric Cober, MD
Advertisement
Advertisement