Locations:

How to spot them with OCT and fundus photography
Inherited retinal dystrophy affects one in 3,000-4,000 people. There are hundreds of genetic subtypes that can cause it, including variations within the gene CRB1, which determines some aspects of the connection between photoreceptors and Müller cells.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
CRB1-related retinal dystrophy — which can manifest as Leber congenital amaurosis in infants, retinitis pigmentosa in teens and young adults, or cone-rod dystrophy at any age — is an uncommon retinal degenerative disease within the rare disease community, says Elias I. Traboulsi, MD, MEd, Director of the Center for Genetic Eye Diseases at Cleveland Clinic. However, today it is being diagnosed more frequently due to an increase in genetic testing.
According to a 2017 article in Ophthalmology, CRB1-related retinal disease is the seventh most common type of inherited retinal disease in the U.S., with 47 new cases diagnosed each year. Other studies around the world suggest it’s even more common. Of the most frequently implicated genes in inherited retinal disease, CRB1 ranks No. 4 in Brazil, No. 3 in the United Arab Emirates and No. 2 in Chile.
“In my clinic, I have a fair number of patients with Leber congenital amaurosis, and the No. 1 responsible gene is CRB1,” says Dr. Traboulsi, who presented this information at the Curing Retinal Blindness (CRB1) Foundation’s global patient summit in July along with Cleveland Clinic ophthalmic genetic counselor Meghan DeBenedictis, MS, CGC, MEd.
DeBenedictis, a member of the CRB1 Foundation’s scientific advisory board, emphasized the importance of genetic testing and genetic counseling for patients.
“Thanks to recent FDA approval of gene therapy for RPE65-related retinal dystrophy, there’s a lot of interest in gene therapy for other types of retinal dystrophies, including CRB1-related disease,” says DeBenedictis. “While we don’t have an FDA-approved CRB1 gene therapy yet, it’s still important that patients get an accurate molecular diagnosis for recurrence risk information, their emotional well-being and their overall medical management. Some inherited retinal diseases are caused by genetic variants — although not in CRB1 — that also can cause systemic health problems.”
Advertisement
Not all genetic variations in CRB1 cause disease. That’s why patients should see a genetic counselor or an eye geneticist who specializes in inherited retinal diseases to accurately order and interpret their genetic test results.
“CRB1-related retinal dystrophy is a recessive disease, meaning you need to inherit a disease-causing mutation from both your mom and your dad,” says DeBenedictis. “If both parents carry a CRB1 mutation, there’s a one-in-four (25%) chance that their child will inherit both mutations and be affected with CRB1-related disease.”
That’s a one-in-four chance per pregnancy, not one in four of their children, she stresses.
“Each child is at risk,” she says. “If carrier parents have four children, all four could have the disease — or none of them could.”
Risk increases when an individual has a CRB1-related disease and their partner also carries a CRB1 mutation. Their children each have a one-in-two (50%) chance of being affected.
“Genetic counselors are experts at these risk assessments and can put them in context for each family,” says DeBenedictis. “By reviewing test results and a family tree, we can explain just how each child, grandchild, sibling and other family member potentially could be impacted by disease.”
CRB1-related retinal dystrophies can become evident and be diagnosed at any age.
“Initially these diseases were categorized under Leber congenital amaurosis,” says Dr. Traboulsi. “Infants with this condition have nystagmus and sometimes extremely poor vision. Their waveforms on electroretinography are flat.”
Advertisement
All retinal dystrophies are bilateral and progressive, he adds. Some patients lose total vision over their lifetime.
In addition to clinical presentation, imaging exams can reveal telltale signs that inform diagnosis.
“Optical coherence tomography (OCT) shows characteristic findings that we don’t see in any other retinal disease,” says Dr. Traboulsi. “Layers of the retina appear thickened and disorganized in a patient with CRB1 disease.”
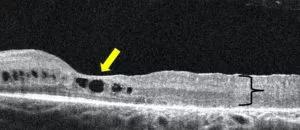
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/5a99b743-982a-4733-b6d8-0f02c2db75e0/20-EYE-1917254-CQD-CRB1-related-retinal-disease-Inset-1-300x130_jpg)
Another characteristic finding is preservation of the para-arteriolar retinal pigment epithelium (RPE). On fundus imaging, this is evident by the darker areas alongside retinal arterioles. These RPE cells also fluoresce on fundus autofluorescence. While not pathognomonic of CRB1 disease, these findings are very suggestive, says Dr. Traboulsi.
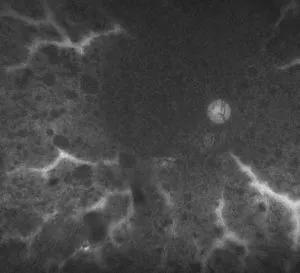
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/d49f1cfc-2a63-4612-86af-2ff486747d91/20-EYE-1917254-CQD-CRB1-related-retinal-disease-Inset-2-300x273_jpg)
A decade ago there was little ophthalmologists could do to treat patients with CRB1-related retinal dystrophy. Today there is hope on the horizon.
The first step for patients is to get genetic testing with an ophthalmic geneticist or genetic counselor subspecialized in ophthalmology.
“Ophthalmic genetic counselors work closely with clinicians and researchers and can keep patients apprised of relevant clinical trials and emerging therapies,” says DeBenedictis.
While gene therapy for CRB1 currently isn’t available, other treatments may be recommended. For example, for cystoid maculopathy, which is common in patients with CRB1 disease, dorzolamide eye drops may help improve vision.
“In addition, we believe there will be a wider window to administer CRB1 gene therapy when it becomes available,” says Dr. Traboulsi. “Natural history data seem to show that retinas aren’t deteriorating as fast as we once thought.”
Advertisement
Advertisement

Motion-tracking Brillouin microscopy pinpoints corneal weakness in the anterior stroma

Registry data highlight visual gains in patients with legal blindness

Prescribing eye drops is complicated by unknown risk of fetotoxicity and lack of clinical evidence

A look at emerging technology shaping retina surgery

A primer on MIGS methods and devices

7 keys to success for comprehensive ophthalmologists

Study is first to show reduction in autoimmune disease with the common diabetes and obesity drugs

Treatment options range from tetracycline injections to fat repositioning and cheek lift