Locations:

Efficacy, safety and tolerability data shared at hematology meeting
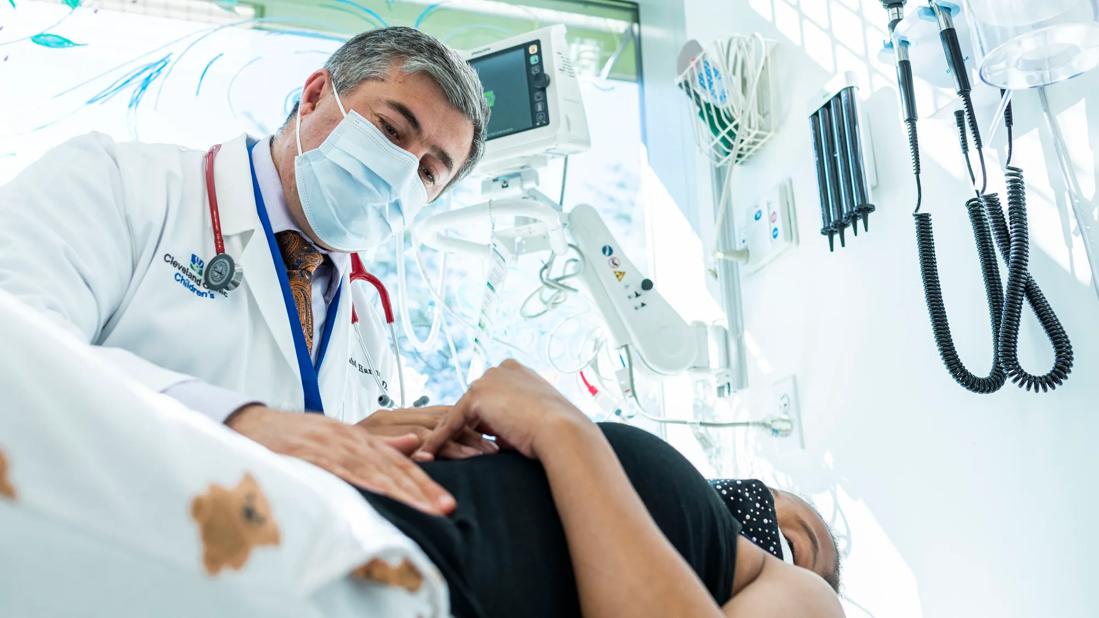
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/a77c9f5a-18d4-4d51-a507-8193cdca17d2/CHP_3261510_09-19-22_141_MLC-jpg)
Clinician looking down at patient on exam table
Results from a gene therapy trial evaluating an experimental treatment continue to show benefits in people with severe sickle cell disease.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Interim findings from RUBY, the multicenter Phase I/II trial, demonstrate preliminary efficacy following infusion of renizgamglogene autogedtemcel (reni-cel), known formally as EDIT-301. The therapy has also shown a favorable safety and tolerability profile.
Rabi Hanna, MD, Director of the Pediatric Bone Marrow Transplantation at Cleveland Clinic Children’s, presented these findings at the 2024 European Hematology Association meeting, the second-largest hematology meeting in the world.
He stresses that while the research team is encouraged by these results, the trial is ongoing.
Sickle cell disease is an inherited blood disorder caused by a single mutation in the hemoglobin gene. It affects more than 20 million people worldwide and 100,000 people in the U.S.
Reni-cel, the investigational, gene-edited, autologous hematopoietic stem cell therapy, could offer potentially a curative option for patients with severe sickle cell disease.
“Using the patient as the source, their own stem cells become the ‘donor’ cells. Then, the advanced gene editing technology, CRISPR/Cas12a, alters the gene in HBG1 and HBG2, mimicking naturally occurring mechanisms involved in the hereditary persistence of fetal hemoglobin (HPFH). This has a protective effect on red blood cells, preventing them from sickling and preventing end-organ damage,” explains Dr. Hanna.
To date, 18 of the study’s 40 enrolled patients have received the reni-cel transfusion. Follow-up ranges from two months to two years, with a median of seven months.
Advertisement
All patients in the study reported being free of a vaso-occlusion event following reni-cel infusion. They also experienced normalization of total hemoglobin and correction of anemia.
Vaso-occlusive pain crises and hemolytic anemia, Dr. Hanna explains, are hallmarks of sickle cell disease.
“In addition to no reported pain crises, patients who received the infusion appear to have normal hemoglobin, which is quite exciting. Because we know that sustained increases to hemoglobin and fetal hemoglobin levels correlate with improved function and better patient-reported outcomes in those with sickle cell disease,” he notes.
Patients achieved early total hemoglobin, with a mean within the normal range at >14 g/dL and improvements in fetal hemoglobin well above levels of >40%.
This sustained improvement in fetal hemoglobin is important, explains Dr. Hanna, “because we know that through the pathology of hemolysis, and not just the sickling, people with sickle cell disease can experience liver and kidney damage,” he says. “So, if we can address that and prevent hemolysis, we may be able to prevent end-organ damage.”
Results also show successful engraftment among all patients who received the infusion. While the study does not directly compare to other gene therapies, the authors observed a relatively early time to engraftment. Time to neutrophil engraftment was a median time of 23.0 days (15.0, 29.0) and time to platelet engraftment was a median time of 24.0 days (18.0, 51.0).
Study data demonstrated favorable safety and tolerability outcomes among all participants. The reni-cel safety profile is consistent with myeloablative busulfan conditioning and autologous hematopoietic stem cell transplantation.
Advertisement
While Dr. Hanna emphasizes these are interim—and not final—results, the data are consistent with the initial findings.
Following screening and consent, patients’ hematopoietic stem and progenitor cells are harvested from peripheral blood. They are given plerixafor, a single-use injection, to mobilize hematopoietic stem cells in preparation for apheresis. The cells are sent to a genome editing company, where they are edited at promoter regions of gamma globin genes 1 and 2 using CRISPR/CASP 12.
Patients in the trial receive myeloablation conditioning with busulfan. This high dose of chemotherapy is important to get rid of the nonfunctional cells and makes room for the modified stem cell infusion. Following the infusion, patients are monitored in the hospital until engraftment.
On the gene editing technology, he says, “CRISPR/Cas12a is an incredibly precise technology that goes directly toward the gene, making edits to the HBG1 and HBG2 promotor regions, reactivating γ-globin, inducing the production of fetal hemoglobin and, ultimately, correcting of anemia,” explains Dr. Hanna.
Patients in the RUBY Trial receive a single infusion of reni-cel with a 24-month follow-up as a primary endpoint.
“It’s been more than 100 years since sickle cell disease was discovered and, up until recently, few options have existed for patients,” Dr. Hanna says.
Allogenic blood or marrow transplant are the only curative options for individuals with the disease, but finding a suitable match is difficult, and outcomes may be suboptimal due to risk of graft rejection and graft-versus-host disease.
Advertisement
Hydroxyurea, first approved by FDA is an antineoplastic drug in the 1960s, was later used for adults with sickle cell disease in the late 1990s. It wasn’t approved for use in pediatric patients with the disease until 2017. While the drug addresses some symptoms and may prevent pain crises in some patients, it’s not curative.
Enrollment for the RUBY trial has concluded, but the study will continue until each patient reaches their two-year follow-up.
Dr. Hanna also remarks that inclusion criteria was amended to include adolescent patients as young as age 12, and the final analysis will reflect data from these patients.
He says that one patient in the study described their experiences with the therapy as freeing: "free from hospitals, free from pain and freeing him to achieve his dreams.”
While the analysis is still underway, the team is hopeful that they will continue to observe positive outcomes in this patient population.
Advertisement
Advertisement

Interim results of RUBY study also indicate improved physical function and quality of life

Integrated care model reduces length of stay, improves outpatient pain management

New course offers insights into clinical, psychosocial and ethical dimensions of care

Watch for sudden unilateral vision loss without pain
Nurses play key role in comprehensive lifetime treatment program

How to combat the rise in mortality when patients become adults

Two-year event-free survival comparable to matched sibling donor myeloablative transplant
First-in-human trials of CRISPR-Cas12a gene editing demonstrate safety and meaningful event-free survival