Locations:

First-in-human trials of CRISPR-Cas12a gene editing demonstrate safety and meaningful event-free survival
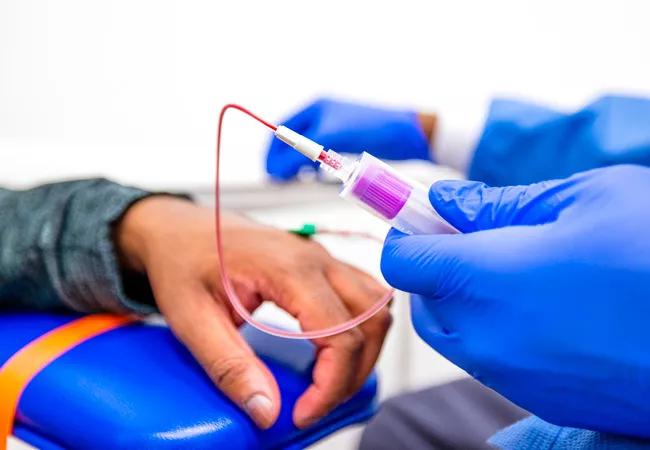
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/6a241202-03f1-4a2d-9683-531a9726b68b/CQD-CHP4445460-hanna-sickle-650x450-1_jpg)
CQD-CHP4445460-hanna-sickle-650×450
Results from the phase 1/2 RUBY and EdiThal trials suggest that CRISPR-Cas12a gene editing has the potential to cure sickle cell disease and transfusion-dependent beta-thalassemia (TDT).
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
The findings, presented in a poster at the 2023 American Society of Hematology conference, indicate the safety and efficacy of infusion of renizgamglogene autogedtemcel (EDIT-301), now called “Reni-Cel,” in 11 patients with severe sickle cell disease and six patients with TDT.
“By three months post-transplant, all of the patients had elevation in their fetal hemoglobin to almost 40%, which has been sufficient to prevent sickling in preclinical trials,” says the study’s first author, Rabi Hanna, MD, Chair of the Department of Pediatric Hematology, Oncology and Bone Marrow Transplant at Cleveland Clinic. “Their markers of hemolysis — lactate dehydrogenase, reticulocyte count, indirect bilirubin and haptoglobin — all normalized. Promising efficacy results also showed that none of the patients with sickle cell disease have experienced vaso-occlusive events and none of the patients with TDT have required transfusions.”
Sickle cell disease and TDT are hereditary genetic blood disorders caused by variants in the HBB gene. In sickle cell disease, a single mutation leads to polymerization of sickle hemoglobin (HbS), causing red blood cells to be sickle-shaped rather than disk-shaped. In TDT, mutation leads to ineffective erythropoiesis and significant anemia.
Both sickle cell disease and TDT are associated with lifelong complications, multi-organ damage and comorbidities that impact quality of life, ultimately leading to a shortened lifespan. The only curative treatment for sickle cell disease and TDT is an allogeneic blood or marrow transplant, but finding a suitable match is often difficult.
Advertisement
EDIT-301 uses novel CRISPR-Cas12a technology to edit HBG1 and HBG2 promoter regions, leading to HbF induction and sustainable red blood cell output. The resulting genomic modifications mimic naturally occurring mutations of hereditary persistence of fetal hemoglobin (HbF) in the gamma-globin gene, reactivating gamma-globin expression and increasing HbF production. Increased HbF levels have been proven to reduce or prevent complications of sickle cell disease and TDT, and they also are associated with decreased incidence of key clinical outcomes, such as stroke and mortality.
First the patient receives bone marrow stimulant plerixafor. Then the patient’s hematopoietic stem cells are harvested from peripheral blood. The cells are sent to Editas Medicine, a genome editing company, where the promoter regions of gamma-globin genes 1 and 2 are edited using CRISPR-Cas12a. Patients are then treated with chemotherapy to make room for the genetically modified cells, which are delivered by infusions. Patients remain in the hospital until they engraft with new, modified stem cells.
“Other commercially available gene therapy products add functional copies of the beta-globin gene, whereas the CRISPR-Cas12a approach is more precise,” explains Dr. Hanna. “It goes directly toward the defect or the targeted gene and cuts it out, so it can be replaced with a gene to activate HbF production.”
The RUBY trial enrolled patients ages 18 to 50 diagnosed with severe sickle cell disease. These trial participants had a history of two or more severe vaso-occlusive events per year in the two years prior to the trial. The primary endpoints were proportion of patients achieving complete resolution of severe vaso-occlusive events and safety and tolerability of EDIT-301.
Advertisement
EdiThal enrolled patients ages 18 to 35 diagnosed with TDT. These trial participants had a history of 100 mgL/kg/year or 10 U/year or more of packed red blood cell transfusions in the two years prior to the trial. The primary endpoints were proportion of participants achieving engraftment and the safety and tolerability of EDIT-301.
After myeloablative conditioning with busulfan, all the patients received a single infusion of EDIT-301. At 24 months, the investigators assessed neutrophil and platelet engraftment, total hemoglobin, HbF, percentage of F-cells, mean HbF concentration/F-cell, markers of hemolysis, transfusion requirement, vaso-occlusive events in sickle cell disease patients and treatment-emergent adverse events.
The data presented at ASH were based on a cutoff of Nov. 22, 2023, for RUBY patients and Nov. 28, 2023, for EdiThal patients.
Compared with a mean of 4.0 severe vaso-occlusive events per year in the two years prior to the trial, none of the 11 sickle cell disease patients in RUBY experienced vaso-occlusive events after treatment with EDIT-301. Mean percentage of HbF was 47.7% by month 4 and was sustained above 40% through last follow-up. By month 5, all sickle cell disease patients had achieved normal hemoglobin levels.
Following EDIT-301 treatment, all five TDT patients in EdiThal maintained hemoglobin levels above 9 g/dL. Transfusion-free intervals for the cohort ranged from 1.8 to 7.5 months, based on having had a last red blood cell transfusion at 0.5 to 2.2 months after receiving EDIT-301.
Advertisement
No serious treatment-emergent adverse events were reported after treatment with EDIT-301. The safety profile of the therapy in patients with sickle cell disease or TDT was consistent with myeloablative conditioning with busulfan and autologous hematopoietic stem cell transplantation.
“We hope to launch a phase 3 trial of EDIT-301 in sickle cell disease next year,” says Dr. Hanna. “If it demonstrates safety and efficacy, CRISPR-Cas12a gene editing may be an equalizer for patients, providing a functional cure that will enable them to pursue their dreams.”
Advertisement
Advertisement

Interim results of RUBY study also indicate improved physical function and quality of life

Integrated care model reduces length of stay, improves outpatient pain management

New course offers insights into clinical, psychosocial and ethical dimensions of care

Watch for sudden unilateral vision loss without pain

Efficacy, safety and tolerability data shared at hematology meeting
Nurses play key role in comprehensive lifetime treatment program

How to combat the rise in mortality when patients become adults

Two-year event-free survival comparable to matched sibling donor myeloablative transplant