Locations:

Who is at risk? What tests should be ordered?

By John A. Morren, MD, and Yuebing Li, MD, PhD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Note: This article is reprinted from the Cleveland Clinic Journal of Medicine (2023;90[2]:103-113). It represents the first portion of the published journal article, with questions pertaining to myasthenia gravis evaluation and diagnosis. Part 2 is focused on questions pertaining to treatment of myasthenia gravis.
The name “myasthenia gravis” comes from the Greek for muscle weakness and the Latin word for grave or serious. A chronic autoimmune neuromuscular disorder causing skeletal muscle weakness, its primary pathophysiology involves dysfunction of the postsynaptic aspect at the neuromuscular junction, mainly a loss of acetylcholine receptor (AChR) function on the muscle membrane.
Certain skeletal muscle groups are more likely to be involved than others, but the pattern varies widely among patients and depends on the clinical course in the individual patient. Accordingly, myasthenia gravis is typically categorized as either ocular (in which weakness is limited to the extrinsic ocular muscles and levator palpebrae superioris) or generalized (in which muscles beyond those in the ocular form are involved, including those of the limbs, the bulbar and oropharyngeal region, and muscles of respiration).
The following frequently asked questions and answers aim to provide up-to-date, clinically relevant information about risk for myasthenia gravis and patient evaluation and diagnosis.
Family members, particularly first-degree relatives, of those with myasthenia gravis have a higher risk not only for myasthenia gravis but also for other autoimmune diseases.1 In addition, the disease has interesting patterns of age, sex and phenotype.
Advertisement
Myasthenia gravis can strike at any age, but the age at onset has a bimodal distribution, with the first peak in patients in their teens and 20s, in which girls and women outnumber boys and men, and the second peak in patients in their 50s and 60s, in which men outnumber women.2,3
In the past, female patients outnumbered male patients overall. However, the age at onset has progressively increased, together with the proportion of men, so that the preponderance of women is becoming less.4,5 There is a male predominance in ocular myasthenia gravis as well.6 Boys and girls are equally affected before puberty, but more girls than boys get the disease afterward.7
The myasthenia gravis subtype possessing antibodies to muscle-specific tyrosine kinase (MuSK) has a marked female predominance (more than 70% in all studies reviewed), and its mean age at onset is 36 to 38 years.2,8
African Americans may have slightly higher rates of myasthenia gravis incidence and prevalence, as well as more severe disease.9,10 In the U.S., 28% to 47% of patients with MuSK antibodies are African American.8 In addition, MuSK antibody-positive myasthenia gravis occurs in a higher proportion of those of Asian ancestry than in those of European or African ancestry.11
About 13% of patients with myasthenia gravis have a comorbid autoimmune disorder.12 Thyroid disease (Hashimoto thyroiditis, Graves’ disease) is the most common, followed by rheumatoid arthritis.12,13 Up to about 10% of patients with myasthenia gravis may have associated thymoma.
Advertisement
Fortunately, myasthenia gravis is uncommon. In a systematic review of 55 studies, Carr et al14 calculated that the pooled incidence was 5.3 per million person-years, and the prevalence was 77.7 per million persons — both considerably lower, for example, than those of hypothyroidism or Guillain-Barré syndrome, which are in the differential diagnosis.
Although the incidence of myasthenia gravis has changed little over time, its estimated prevalence has significantly increased since the 1950s, mostly owing to improvements in diagnosis and treatment that have reduced the mortality rate so that more people are living with the disease.
Think about myasthenia gravis when a patient has fatigable weakness, especially weakness of ocular muscles producing variable diplopia, ptosis and weak eye closure. These are the core clinical features. At initial presentation, which is typically subacute, up to 85% of patients have ocular symptoms.15
Fatigable is key. The muscle weakness fluctuates, classically worsening with sustained or repetitive physical activity, worsening by evening or nighttime, and improving with rest. In the arms and legs, the weakness generally tends to affect proximal muscles more than distal ones. In the mouth and neck, prominent bulbar weakness, including dysarthria, nasal speech, dysphagia, poor saliva control, difficulty chewing and neck weakness (including a dropped head phenotype) may be seen in about 15% of patients at presentation.15 Myasthenia gravis-related weakness may progress in severity over weeks or months, often with exacerbations and remissions during its course.
Advertisement
Of importance, patients with myasthenia gravis typically have no sensory or pain symptoms, bowel or bladder dysfunction, or changes in mental status or cognition. In addition, deep tendon reflexes are usually intact, even if the patient has marked weakness.
The table below lists common disorders in the differential diagnosis of myasthenia gravis and their distinguishing features.
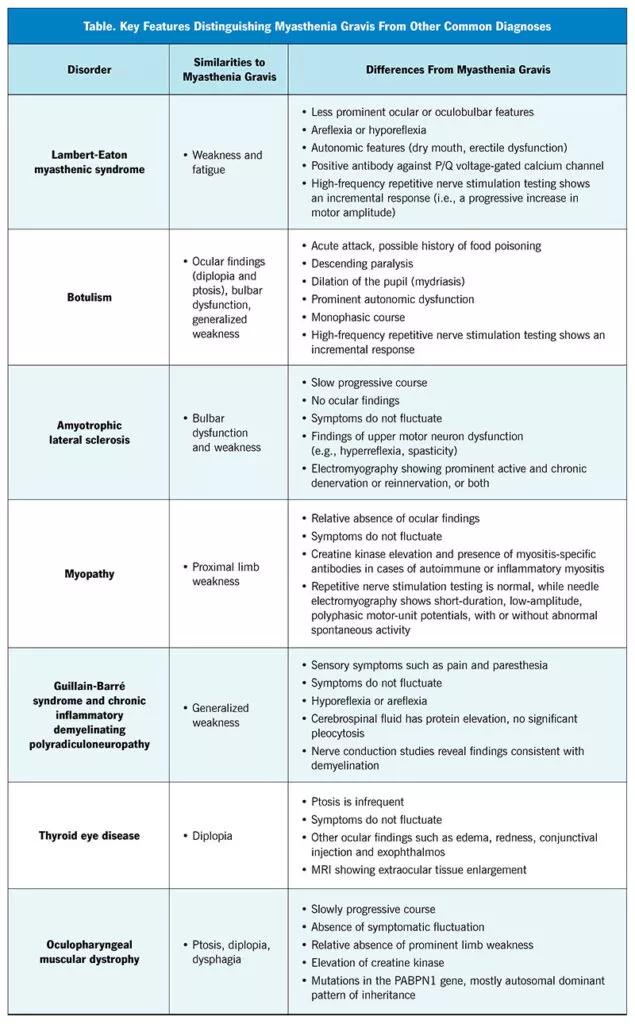
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/a9fdf203-fd50-48a2-8bb3-6d902b10dd8c/23-NEU-3863417-CQD-Inset-First-Table-800x1290-1-635x1024_jpg)
First-line diagnostic tests are typically serologic.
Antibody tests
Anti-AChR antibody(particularly the binding subtype) is highly specific (> 90%) and very sensitive (up to about 85%) in those with generalized myasthenia gravis.2
Anti-MuSK antibodies.In patients with myasthenia gravis who are seronegative for anti-AChR antibodies, up to 37% possess anti-MuSK antibodies.8 However, the sensitivity of anti-AChR antibody is lower, about 50%, in those who have purely ocular myasthenia gravis. Anti-MuSK antibodies rarely occur in the group of patients with purely ocular myasthenia gravis.15
Antilipoprotein-related protein 4 (LRP4) antibodyis found in 3% to 50% of the remaining patients with generalized myasthenia gravis who are seronegative to both anti-AChR and anti-MuSK antibodies.
Antistriated muscle antibodies.On immunofluorescent staining, antistriated muscle antibodies bind in a cross-striational pattern to a number of muscle proteins including titin, ryanodine receptor, actin, myosin, tropomyosin and filamin. They are much less specific for myasthenia gravis and are seen in about 30% of patients, and they are more useful as a marker for thymoma, especially in the nonelderly.15 Thus, myasthenia gravis cannot be reliably diagnosed on the basis of positive antistriated muscle antibody alone.
Advertisement
Electrodiagnostic tests
Two electrodiagnostic tests — repetitive nerve stimulation and single-fiber electromyography — provide objective evidence of impairment of neuromuscular junction transmission and are helpful in diagnosing myasthenia gravis. They need not be performed in all patients, but they provide supportive diagnostic evidence, especially in seronegative patients and when prompt confirmation of the diagnosis is needed.
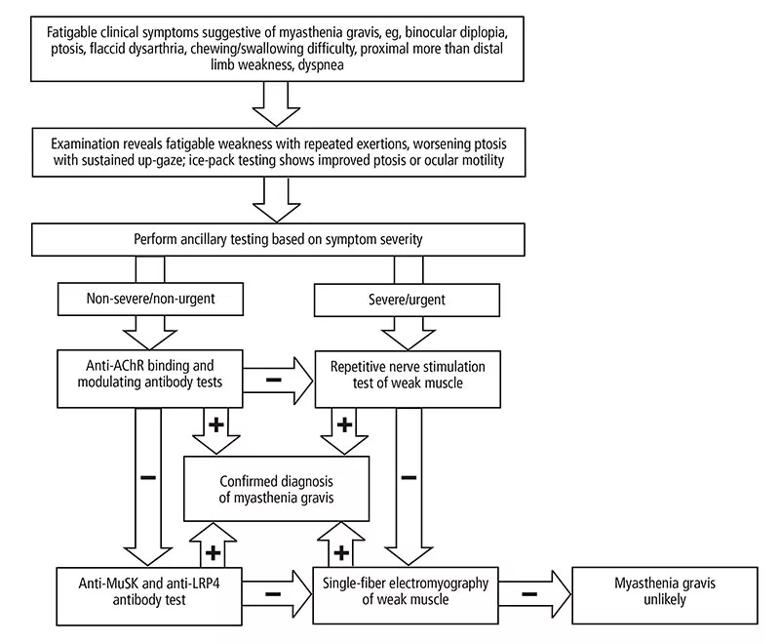
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/b406be49-332d-4e4a-95fa-44e573447c5f/23-NEU-3863417-CQD-Inset-800x660-1_jpg)
Figure 1. Diagnostic algorithm for myasthenia gravis. If the anti-AChR binding and modulating antibody tests are negative, two options are reasonable, as indicated. AChR = acetylcholine receptor; LRP4 = lipoprotein-related protein 4; MuSK = muscle-specific tyrosine kinase
Repetitive nerve stimulationuses repeated “trains” of nerve stimulation to generate electrical muscle responses. The amplitudes of these responses can be measured to gauge the fatigability of neuromuscular junction transmission. The sensitivity and specificity of repetitive nerve stimulation depends on the nerve-muscle combinations examined, the severity of myasthenia gravis, and the cutoff values used for a decremental response. Its overall diagnostic sensitivity ranges from about 30% to 80% for generalized myasthenia gravis, with lower sensitivity in milder disease or when distal muscles are tested. In ocular myasthenia gravis, its sensitivity is only 10% to 30%.16
Single-fiber electromyographyuses small needle electrodes to measure the variability of single muscle fiber potentials, a reflection of neuromuscular junction transmission. This test is often considered only when other diagnostic tests are unrevealing. It is more sensitive than repetitive nerve stimulation (62% to 99% for ocular myasthenia gravis, and 75% to 98% for generalized myasthenia gravis). Thus, a normal result in a clinically weak muscle essentially rules out myasthenia gravis. Its reported specificity varies from 66% to 98% for ocular myasthenia gravis and up to 98% for generalized myasthenia gravis, and abnormal results can be seen in other neuromuscular disorders such as motor neuron disease, congenital myasthenia gravis or myopathy.17
Other tests
Also useful in patients suspected of having myasthenia gravis are tests for common comorbid conditions, e.g., chest CT or MRI for thymic abnormalities. One should be alert for clinical features that may suggest comorbid autoimmune conditions that would call for additional serologic tests, such as thyroid-stimulating immunoglobulin, antithyroid peroxidase, antithyroglobulin or rheumatoid factor.
1. Liu FC, Kuo CF, See LC, Tsai HI, Yu HP. Familial aggregation of myasthenia gravis in affected families: a population-based study. Clin Epidemiol. 2017;9:527-535. doi:10.2147/CLEP.S146617
2. Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve. 2004;29(4):484-505. doi:10.1002/mus.20030
3. Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37(2):141-149. doi:10.1002/mus.20950950
4. Pakzad Z, Aziz T, Oger J. Increasing incidence of myasthenia gravis among elderly in British Columbia, Canada. Neurology. 2011;76(17):1526-1528. doi:10.1212/WNL.0b013e318217e735
5. Sanders DB, Raja SM, Guptill JT, Hobson-Webb LD, Juel VC, Massey JM. The Duke myasthenia gravis clinic registry: I. Description and demographics. Muscle Nerve. 2021;63(2):209-216. doi:10.1002/mus.27120
6. Nair AG, Patil-Chhablani P, Venkatramani DV, Gandhi RA. Ocular myasthenia gravis: a review. Indian J Ophthalmol. 2014;62(10):985-991. doi:10.4103/0301-4738.145987
7. Finnis MF, Jayawant S. Juvenile myasthenia gravis: a paediatric perspective. Autoimmune Dis. 2011;2011:404101. doi:10.4061/2011/404101
8. Morren J, Li Y. Myasthenia gravis with muscle-specific tyrosine kinase antibodies: a narrative review. Muscle Nerve. 2018;58(3):344-358. doi:10.1002/mus.26107
9. Phillips LH 2nd, Torner JC, Anderson MS, Cox GM. The epidemiology of myasthenia gravis in central and western Virginia. Neurology. 1992;42(10):1888-1893. doi:10.1212/wnl.42.10.1888
10. Oh SJ, Morgan MB, Lu L, et al. Racial differences in myasthenia gravis in Alabama. Muscle Nerve. 2009;39(3):328-332. doi:10.1002/mus.21191
11. Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia gravis: epidemiology, pathophysiology and clinical manifestations. J Clin Med. 2021;10(11):2235. doi:10.3390/jcm10112235
12. Mao ZF, Yang LX, Mo XA, et al. Frequency of autoimmune diseases in myasthenia gravis: a systematic review. Int J Neurosci. 2011;121(3):121-129. doi:10.3109/00207454.2010.539307
13. Christensen PB, Jensen TS, Tsiropoulos I, et al. Associated autoimmune diseases in myasthenia gravis. A population-based study. Acta Neurol Scand. 1995;91(3):192-195. doi:10.1111/j.1600-0404.1995.tb00432.x
14. Carr AS, Cardwell CR, McCarron PO, McConville J. A systematic review of population based epidemiological studies in myasthenia gravis. BMC Neurol. 2010;10:46. doi:10.1186/1471-2377-10-46
15. Beloor Suresh A, Asuncion RMD. Myasthenia gravis. In: StatPearls. Treasure Island, Florida: StatPearls Publishing; December 15, 2021.
16. Lamb CJ, Rubin DI. Sensitivity and specificity of repetitive nerve stimulation with lower cutoffs for abnormal decrement in myasthenia gravis. Muscle Nerve. 2020;62(3):381-385. doi:10.1002/mus.26999
17. Morren JA, Levin KH, Shields RW. Diagnostic accuracy of single fiber electromyography for myasthenia gravis in patients followed longitudinally. J Clin Neurophysiol. 2016;33(5):469-474. doi:10.1097/WNP.0000000000000285
Dr. Morren and Dr. Li are both staff neurologists in the Neuromuscular Center in Cleveland Clinic’s Neurological Institute.
Advertisement

An overview of associated antibodies, therapies for antibody-positive disease and the outlook for atypical forms of MG

Retrospective analysis assesses frequency and severity relative to a reference antibiotic
Guidance on instructing patients, treatment selection, impact of disease course and more

Guidance for the 5% to 20% of patients with suboptimal response or tolerability

Aim is for use with clinician oversight to make screening safer and more efficient

Rapid innovation is shaping the deep brain stimulation landscape

Study shows short-term behavioral training can yield objective and subjective gains

How we’re efficiently educating patients and care partners about treatment goals, logistics, risks and benefits