Locations:

Guidance on instructing patients, treatment selection, impact of disease course and more
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/bc0d122f-5d49-4b37-be00-e69dd763e13b/23-NEU-3941958-neuro-exam-650x450-1_jpg)
23-NEU-3941958-neuro-exam-650×450
By John A. Morren, MD, and Yuebing Li, MD, PhD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Note: This article is reprinted from the Cleveland Clinic Journal of Medicine (2023;90[2]:103-113). It represents the second portion of the published journal article, with questions pertaining to the treatment of myasthenia gravis. Part 1 is focused on questions pertaining to evaluation and diagnosis of myasthenia gravis.
The name “myasthenia gravis” comes from the Greek for muscle weakness and the Latin word for grave or serious. A chronic autoimmune neuromuscular disorder causing skeletal muscle weakness, its primary pathophysiology involves dysfunction of the postsynaptic aspect at the neuromuscular junction, mainly a loss of acetylcholine receptor (AChR) function on the muscle membrane. Myasthenia gravis is typically categorized as either ocular (in which weakness is limited to the extrinsic ocular muscles and levator palpebrae superioris) or generalized (in which muscles beyond those in the ocular form are involved).
The following frequently asked questions and answers aim to provide up-to-date, clinically relevant information about myasthenia gravis.
Myasthenia gravis tends to progress, especially in the first several years, so we recommend treating it aggressively with immunosuppressants at the outset and then gradually easing back.
Not until the late 1960s was myasthenia gravis recognized as an immune-mediated disorder, and immunotherapies such as corticosteroids, azathioprine and methotrexate started to be used as treatments for it.1 As a result, studies of its outcome done before the late 1960s generally reflected its natural course. In several such early studies, the mortality and morbidity rates were highest within the first three years of the disease and lower thereafter.2-4
Advertisement
In particular, ocular myasthenia gravis reaches its maximal severity within the first three years in most patients.4 In older studies, approximately two-thirds of cases of ocular myasthenia gravis subsequently progressed into the generalized subtype; of these, approximately 80% did so within the first year and 90% within the first three years.4,5 In more recent series, the percentage of generalization from the ocular subtype was less, as low as 20%6 to 50%.7
More immunotherapies for myasthenia gravis are now available. However, the aforementioned studies of the natural course help guide the assessment of risks and benefits of immunosuppressive treatment. While the early goal should focus on aggressive treatment to improve the patient’s functional status, care must be taken to avoid serious adverse effects from intense immunotherapy. Patients who endure the first three years with relatively good symptom control tend to have a higher chance of gradual improvement or a steady state and, less often, worsening of the disease.4,8,9 An exception is in refractory myasthenia gravis, which accounts for approximately 10% of patients with generalized myasthenia gravis and can be associated with relapses and exacerbations late in the course.
In the long term, it is preferable to steadily minimize immunosuppression if the patient’s condition remains stable, while watching for relapse or exacerbation. Approximately half of patients can achieve remission or minimal symptoms with low-dose immunotherapy.9 However, clinicians should be cautious about discontinuing immunotherapy completely, as only about 10% of patients may achieve complete stable remission off immunotherapy.10
Advertisement
After myasthenia gravis is diagnosed, patients should be educated about its typical course and largely benign prognosis. Points to discuss include:
Many patients with myasthenia gravis are cautious about physical exertion, fearing that exercise might worsen their symptoms. However, most can tolerate and benefit from some form of exercise. Patients with mild disease can participate in resistance and aerobic training. For those with severe symptoms, stretching exercises such as tai chi, yoga and balance training are usually most appropriate. Simply being more active and reducing overall sedentary time are important.12
Fatigue is common, reported in approximately 80% of patients at some stage of their disease. It is important to recognize differences between fatigue and fatigable weakness, as fatigue does not call for escalating myasthenia gravis treatment. The cause of fatigue in myasthenia gravis is multifactorial and includes deconditioning, cognitive blunting, sleep disturbance, and weight gain. Management of fatigue may include regular exercise, sleep evaluation, psychotherapy and cognitive behavioral therapy.12
Advertisement
Because some medications can trigger or worsen myasthenic symptoms, all patients with myasthenia gravis, especially those with significant weakness, should be observed for increased weakness whenever a new medication is started. In principle, if a patient’s condition deteriorates when given a new drug, the drug should be withdrawn. Drugs that are most clearly contraindicated in myasthenia gravis include telithromycin, intravenous magnesium, botulinum toxin, penicillamine and immune checkpoint inhibitors (see discussion below).13,14
Other medications that can worsen the disease include fluoroquinolones, macrolide antibiotics, aminoglycosides, beta-blockers, chloroquine, statins and iodinated contrast (mostly associated with a low overall risk of aggravating myasthenia gravis). Most patients with mild to moderate disease or in stable remission tolerate these drugs without ill effect. Some medications (e.g., aminoglycosides) are probably best avoided, as many alternatives are available. More robust data are needed to ascertain and quantify the risk of myasthenia gravis worsening with the other medications mentioned above.
Immune checkpoint inhibitors, used to treat malignancies, have become the most common iatrogenic cause of myasthenia gravis. They include blockers of programmed cell death receptor 1 (nivolumab, pembrolizumab), programmed cell death ligand 1 (atezolizumab, durvalumab, and avelumab) and cytotoxic T cell lymphocyte-associated antigen 4 (ipilimumab).
Immune checkpoint inhibitors can exacerbate symptoms in patients with myasthenia gravis or cause de novo disease. Many patients who develop myasthenia gravis as a result of these drugs have elevations of creatine kinase and troponin due to coexisting necrotizing myositis and myocarditis. The range of these autoimmune complications is wide — mild and monophasic in some patients, fulminant and even fatal in others. Prompt recognition is critical, as the immune checkpoint inhibitor needs to be stopped promptly and immunotherapy added.15
Advertisement
Pyridostigmine, the most commonly used acetylcholinesterase inhibitor for symptomatic treatment of myasthenia gravis, is typically used alone in mild cases or in combination with immunosuppressants in more severe ones. However, its efficacy may be minimal in patients with long-standing or severe myasthenia gravis.
Pyridostigmine’s onset of effect is 30 to 60 minutes after each dose, and its duration is three to six hours. It should be taken 30 minutes before meals if dysphagia is present. A typical starting dose is 60 mg every six hours during daytime.
Patients who awaken with morning weakness can take a 180-mg extended-release formulation before sleep. However, the response to this formulation varies due to erratic absorption.
The dosage of pyridostigmine can be titrated up to 240 to 360 mg daily, but side effects are more common at higher doses, and overdose may result in increased weakness.16 In practice, if a patient needs more than 240 mg per day, it is time to move on to immunotherapy.
Once myasthenia gravis is controlled with immunotherapy, most patients do not need pyridostigmine. In a 1973 study in animals, long-term acetylcholinesterase inhibitor treatment at high doses led to degeneration and dysfunction of the neuromuscular junction,17 but clinical experience suggests that pyridostigmine is generally safe without significant long-term complications.
The most common side effects are gastrointestinal (e.g., abdominal cramping, loose stool and flatulence). Bradycardia, bronchospasm, increased sweating, excessive lacrimation, muscle twitching and cramping are other effects.
To manage side effects, oral glycopyrrolate or hyoscyamine can be taken concurrently with pyridostigmine doses. Dosage adjustment may be required in patients with renal impairment. One should be vigilant for the development of bronchospasm in patients with asthma.
Patients with MuSK antibody-positive myasthenia gravis may not respond well to pyridostigmine or may develop profuse cramps and fasciculations, even with low doses, possibly owing to reduction of cholinesterase levels at the neuromuscular junction.
According to consensus guidelines,13 corticosteroids or nonsteroidal immunosuppressive drugs should be used in all patients with myasthenia gravis who have not met their treatment goals after an adequate trial of pyridostigmine.
Only two controlled trials have evaluated the efficacy of corticosteroids in generalized myasthenia gravis.18,19 However, retrospective studies of oral steroids (prednisone or prednisolone) as the main myasthenia gravis treatment also provide evidence that these drugs are effective.20 Corticosteroids help nearly all patients with all subtypes of myasthenia gravis, resulting in marked improvement in more than 80%. Their onset of action is relatively rapid (two weeks, on average).
Outpatients with mild to moderate symptoms can start prednisone at 20 mg daily and gradually increase the daily dose by 10 mg every one to two weeks up to approximately 60 mg daily, titrating to clinical response. Other corticosteroids with proven efficacy in myasthenia gravis include methylprednisolone, given intramuscularly or intravenously, and oral dexamethasone.20
Some patients respond better than others to corticosteroids. Good responders have a smooth and consistent response to moderate or high corticosteroid doses and can be kept in remission with low doses (e.g., 5 to 7.5 mg of prednisone daily) without need for nonsteroidal immunosuppressive agents. The long-term risk of such low-dose prednisone therapy is considered minimal.21 Data suggest that patients over age 40, and especially those over age 60, are more likely to be good responders compared with younger patients.20
When starting corticosteroids, be alert for corticosteroid “dipping,” i.e., an exacerbation in myasthenic symptoms, seen in up to half of patients and usually occurring within the first week of starting treatment. Most cases are mild, and the worsening does not lead to the need for intubation or assisted ventilation. Dipping does not predict a poor long-term response to corticosteroid therapy.22 Titrating the dose upward more gradually appears to reduce the occurrence of corticosteroid dipping.23
Once significant improvement is seen after starting corticosteroid therapy, there is no need to wait for maximum improvement to occur before starting to taper these drugs. Weaning should be slow and usually starts after several weeks of high-dose therapy. Initial steroid tapering typically involves reducing the daily dose of prednisone by about 5 to 10 mg per month.
Nonsteroidal immunosuppressive therapies should be considered in the following situations:
Nonsteroidal immunosuppressive drugs such as azathioprine, mycophenolate mofetil, methotrexate, cyclosporine, tacrolimus and rituximab have been extensively used in myasthenia gravis to spare the use of corticosteroids in some patients. Newer agents recently approved, such as eculizumab, ravulizumab and efgartigimod, could also serve this purpose in selected patients.24-26
Other factors, such as antibody status, comorbidities, desired time course of action, and physician or patient preference, may modify the choice of nonsteroidal immunosuppressive therapy. Rituximab is particularly effective for MuSK antibody-positive myasthenia gravis. Azathioprine, methotrexate and mycophenolate mofetil may take six to 12 months to work, while the onset of action of cyclosporine, tacrolimus and rituximab is generally quicker. Several of these drugs can damage the bone marrow, liver, kidneys and lungs, and the functional status of these organs may influence their use.27
At times, nonsteroidal immunosuppressive therapy may also be given as the initial immunosuppressant for patients with mild disease who are content with a slow course of improvement. In patients with significant weakness who have contraindications to corticosteroids, intravenous immunoglobulin, efgartigimod or plasmapheresis can be used in the beginning to expedite clinical improvement while allowing time for an alternative nonsteroidal immunosuppressive therapy to produce its therapeutic effect.28
Because of the delayed action of some nonsteroidal immunosuppressive therapies, prednisone should be started concurrently. In general, however, one should avoid combining more than two immunosuppressants (e.g., prednisone and a nonsteroidal immunosuppressive drug) in view of increased risks of infection and other side effects. An exception is in refractory myasthenia gravis, which often requires intense immunotherapy with multiple agents.29
For patients who gain good control by taking the combination of prednisone and a nonsteroidal immunosuppressive drug, prednisone is usually tapered first. After prednisone is tapered off or reduced to an acceptable minimal dose, the nonsteroidal drug can be tapered next, but much more slowly, usually over years. In some patients, both prednisone and the nonsteroidal drug can be kept at low dosages for optimal disease control and to minimize the side effects of each while taking advantage of their different mechanisms of action.
The thymus gland is essential in the development of central tolerance and T-cell differentiation, and thus likely plays an important role in the immunopathogenesis of myasthenia gravis.
In approximately 10% of patients, myasthenia gravis is a paraneoplastic manifestation of an underlying thymic neoplasm (usually thymoma, rarely thymic carcinoma). However, thymic lymphoid hyperplasia is seen in up to 65% of patients with myasthenia gravis.30 Lymphoid hyperplasia consists of numerous lymphocytes, macrophages and plasma cells, reflecting the autoimmunity underlying myasthenia gravis that often begins in the thymus gland.
There is also evidence to suggest that autoimmunity against the AChR may be due to intrathymic “myoid” cells and medullary thymic epithelial cells that elaborate the AChR or subunits of it on their cell surface.31
The decision to remove the thymus is often influenced by whether patients have thymomatous or nonthymomatous myasthenia gravis. Thymectomy is indicated in all patients with thymic neoplasms. Otherwise, candidacy for thymectomy depends on several factors, including AChR antibody status, myasthenia gravis type, disease duration and patient age.
Supportive evidence comes from the landmark Thymectomy Trial in Non-Thymomatous Myasthenia Gravis Patients Receiving Prednisone Therapy (MGTX).32 To enter that trial, patients had to meet the following criteria:
Thymectomy in similar adult patients age 50 or younger is likely to improve clinical outcomes and permit minimal pharmacotherapy, including immunosuppressant use and dosage.
The benefit of thymectomy in patients ages 51 to 65 is more equivocal, and thymectomy is generally avoided in patients over age 65, since the risk-to-benefit ratio is less favorable.
There is no significant evidence to support thymectomy in those with MuSK antibody-positive myasthenia gravis. However, most experts would also consider thymectomy for patients with generalized myasthenia gravis who are “triple seronegative” (without antibodies to AChR, MuSK or LRP4). This appears to be supported by evidence of similar benefits in both AChR antibody-positive and AChR antibody-negative myasthenia gravis subgroups.33 Thymectomy for patients with strictly ocular myasthenia gravis is controversial.
Although the surgery employed in the MGTX trial was traditional extended transsternal thymectomy via a median sternotomy, this has largely been replaced by less invasive procedures including video-assisted and robotically assisted thymectomy via a transthoracic approach, and extended transcervical thymectomy through a low horizontal neck incision. Retrospective studies have shown similar clinical outcomes from the different surgical techniques.34,35 The major advantages of less invasive surgical approaches relate to their lower postoperative complication rates and shorter hospital stay.
A myasthenic crisis is a life-threatening worsening of myasthenia gravis-related respiratory or bulbar muscle weakness that is severe enough to require intubation, mechanical ventilation or both.13 If a patient has marked dysphagia, managing saliva and other oropharyngeal secretions can become difficult and the risk of aspiration is high.
Key measures in preventing myasthenic crisis are consistent disease control (including adherence to the medication regimen and careful weaning from immunosuppressants) and avoiding triggers or precipitants.
Recognizing myasthenic crisis. Most patients with myasthenic crisis do not present with respiratory insufficiency alone. Rather, neuromuscular respiratory weakness usually occurs in the context of already worsening generalized or bulbar weakness, or both. Therefore, clinical features indicating significant worsening deficits in these areas may provide warning signs.
Of note, classic features of respiratory distress, such as use of accessory muscles of respiration, may be blunted during a myasthenic crisis, so these should not be overly relied upon. Orthopnea is a more specific feature than dyspnea, indicating significant neuromuscular respiratory weakness (especially of the diaphragm). Significant weakness in neck flexors and shoulder external rotators also typically correlates with respiratory muscle weakness.36
A screening test that can be done at the bedside or over the telephone is the single-breath counting test.37 The patient is asked to take a deep inspiration and, on subsequent expiration, count from 1 onwards at a routine speaking pace (about two counts per second) until they need to take another breath. Inability to count to 20 with a single breath indicates significant respiratory weakness.
However, more formal spirometric measures are ideal, and the “20-30-40 rule” should be kept in mind.36 This means that patients should be admitted or transferred to the intensive care unit for airway and respiratory management if vital capacity falls below 20 mL/kg, if the maximal inspiratory pressure (also known as negative inspiratory force) becomes less negative than −30 cm H2O, or if the maximal expiratory pressure falls below 40 cm H2O. Intensive care may also be warranted if the values are falling quickly (> 30% over 24 hours). It is very important that spirometry be done with a well-fitting face mask instead of a mouthpiece when there is significant weakness of facial muscles (particularly orbicularis oris), causing poor seal.
Measures of oxygenation, including pulse oximetry and arterial partial pressure of oxygen, are less helpful than those for carbon dioxide retention because of the prevailing mechanism of ventilatory compromise.
Managing myasthenic crisis.Managing myasthenic crisis entails optimizing medical management of intercurrent medical illness (including infections), removing any culprit medications and giving aggressive immunotherapies aimed at quickly improving neuromuscular junction transmission.
The main therapies are plasmapheresis (also known as plasma exchange) and intravenous immunoglobulin, but usually not both. Both plasmapheresis and intravenous immunoglobulin may begin to produce clinical improvements within several days. However, since their efficacy may start to wane within a few weeks, concomitant augmentation of baseline immunotherapy (e.g., corticosteroids) is needed. Anticholinesterase medications are generally withheld during a myasthenic crisis, especially if the patient has to be intubated, since discontinuation will reduce oropharyngeal secretions and aspiration risk.
Although general principles of weaning and extubation apply to those intubated and mechanically ventilated for myasthenic crisis, one should be mindful of more specific considerations. In particular, there should be a consistent reassuring trend in oropharyngeal secretion clearance and pulmonary function parameters (vital capacity > 15 to 20 mL/kg, maximal inspiratory pressure more negative than –25 to –30 cm H2O) before weaning and attempted extubation. The best approach utilizes daily spontaneous breathing trials after initiation of intravenous immunoglobulin or plasmapheresis treatment.38 Persistent neck flexor weakness may indicate a lower likelihood of successful extubation.
The complement inhibitors eculizumab and ravulizumab and the neonatal Fc-receptor blocker efgartigimod have been recently approved by the FDA for treating AChR antibody-positive myasthenia gravis, and many newer treatments with various mechanisms of action are being studied (Table). Several of them (including rozanolixizumab and zilucoplan) have had positive results in phase 3 trials.39
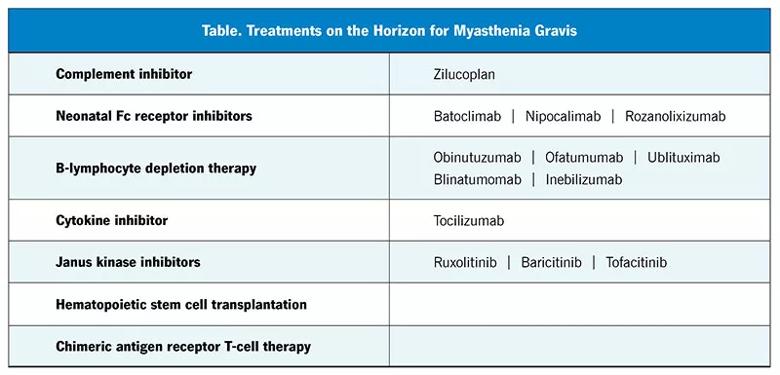
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/e2c3b4bb-e980-4577-a5ea-437f07097b10/23-NEU-3941958-Table-800x385-1_jpg)
The newer immunotherapies are generally more selective in their immunologic targets than the older ones. Accordingly, they have the advantage of causing fewer adverse effects, including life-threatening infections. However, they are very expensive, and a major drawback is their “financial toxicity.” For many patients, the older broad-spectrum immunotherapies will remain a key component of treatment due to lower cost, ease of use and potential of inducing remission. Nonetheless, the pace of major therapeutic innovations in the field is unprecedented, and the future of myasthenia gravis treatment is promising.
1. Keesey J, Aarli J. Something in the blood? A history of the autoimmune hypothesis regarding myasthenia gravis. J Hist Neurosci. 2007;16(4):395-412. doi:10.1080/09647040600675322
2. Simpson JA. An evaluation of thymectomy in myasthenia gravis. Brain. 1958;81(1):112-144. doi:10.1093/brain/81.1.112
3. Oosterhuis HJ. The natural course of myasthenia gravis: a long term follow up study. J Neurol Neurosurg Psychiatry. 1989;52(10):1121-1127. doi:10.1136/jnnp.52.10.1121
4. Grob D, Arsura EL, Brunner NG, Namba T. The course of myasthenia gravis and therapies affecting outcome. Ann N Y Acad Sci. 1987;505:472-499. doi:10.1111/j.1749-6632.1987.tb51317.x
5. Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci. 2004; 217(2):131-133. doi:10.1016/j.jns.2003.08.017
6. Mazzoli M, Ariatti A, Valzania F, et al. Factors affecting outcome in ocular myasthenia gravis. Int J Neurosci. 2018;128(1):15-24. doi:10.1080/00207454.2017.1344237
7. Peeler CE, De Lott LB, Nagia L, Lemos J, Eggenberger ER, Cornblath WT. Clinical utility of acetylcholine receptor antibody testing in ocular myasthenia gravis. JAMA Neurol. 2015;72(10):1170-1174. doi:10.1001/jamaneurol.2015.1444
8. Mantegazza R, Beghi E, Pareyson D, et al. A multicentre follow-up study of 1152 patients with myasthenia gravis in Italy. J Neurol. 1990;237(6):339-344. doi:10.1007/BF00315656
9. Salins S, Teter B, Kavak K, Wolfe GI, Silvestri NJ. Low-dose medication and long-term outcome in myasthenia gravis. J Clin Neuromuscul Dis. 2016;18(2):61-66. doi:10.1097/CND.0000000000000122
10. Sanders DB, Evoli A. Immunosuppressive therapies in myasthenia gravis. Autoimmunity. 2010;43(5-6):428-435. doi:10.3109/08916930903518107
11. Gummi RR, Kukulka NA, Deroche CB, Govindarajan R. Factors associated with acute exacerbations of myasthenia gravis. Muscle Nerve. 2019;60(6):693-699. doi:10.1002/mus.26689
12. O’Connor L, Westerberg E, Punga AR. Myasthenia gravis and physical exercise: a novel paradigm. Front Neurol. 2020;11:675. doi:10.3389/fneur.2020.00675
13. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology. 2016;87(4):419-425. doi:10.1212/WNL.0000000000002790
14. Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96(3):114-122. doi:10.1212/WNL.0000000000011124
15. Dubey D, David WS, Reynolds KL, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing spectrum. Ann Neurol. 2020;87(5):659-669. doi:10.1002/ana.25708
16. Maggi L, Mantegazza R. Treatment of myasthenia gravis: focus on pyridostigmine. Clin Drug Investig. 2011;31(10):691-701. doi:10.2165/11593300-000000000-00000
17. Engel AG, Lambert EH, Santa T. Study of long-term anticholinesterase therapy. Effects on neuromuscular transmission and on motor end-plate fine structure. Neurology. 1973;23(12):1273-1281. doi:10.1212/wnl.23.12.1273
18. Howard FM Jr, Duane DD, Lambert EH, Daube JR. Alternate-day prednisone: preliminary report of a double-blind controlled study. Ann N Y Acad Sci. 1976;274:596-607. doi:10.1111/j.1749-6632.1976.tb47718.x
19. Lindberg C, Andersen O, Lefvert AK. Treatment of myasthenia gravis with methylprednisolone pulse: a double blind study. Acta Neurol Scand. 1998;97(6):370-373. doi:10.1111/j.1600-0404.1998.tb05968.x
20. Morren J, Li Y. Maintenance immunosuppression in myasthenia gravis, an update. J Neurol Sci. 2020;410:116648. doi:10.1016/j.jns.2019.116648
21. Utsugisawa K, Suzuki S, Nagane Y, et al. Health-related quality-of-life and treatment targets in myasthenia gravis. Muscle Nerve. 2014;50(4):493-500. doi:10.1002/mus.24213
22. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15(3):291-298. doi:10.1002/ana.410150316
23. Seybold ME, Drachman DB. Gradually increasing doses of prednisone in myasthenia gravis. Reducing the hazards of treatment. N Engl J Med. 1974;290(2):81-84. doi:10.1056/NEJM197401102900204
24. Howard JF Jr, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study [published correction appears in Lancet Neurol. 2017;16(12 ):954]. Lancet Neurol. 2017;16(12):976-986. doi:10.1016/S1474-4422(17)30369-1
25. Vu T, Meisel A, Mantegazze R, et al. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. 2022;1(5). doi:10.1056/EVIDoa2100066
26. Howard JF Jr, Bril V, Vu T, et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, phase 3 trial [published correction appears in Lancet Neurol. 2021;20(8):e5]. Lancet Neurol. 2021;20(7):526-536. doi:10.1016/S1474-4422(21)00159-9
27. Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018;36(2):311-337. doi:10.1016/j.ncl.2018.01.011
28. Nagane Y, Suzuki S, Suzuki N, Utsugisawa K. Early aggressive treatment strategy against myasthenia gravis. Eur Neurol. 2011;65(1):16-22. doi:10.1159/000322497
29. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies [published correction appears in Ther Adv Neurol Disord. 2018;11:1756286418765591]. Ther Adv Neurol Disord. 2018;11:1756285617749134. doi:10.1177/1756285617749134
30. Nishino M, Ashiku SK, Kocher ON, et al. The thymus: a comprehensive review [published correction appears in Radiographics. 2017;37(3):1004]. Radiographics. 2006;26(2):335-348. doi:10.1148/rg.262045213
31. Hohlfeld R, Wekerle H. Reflections on the “intrathymic pathogenesis” of myasthenia gravis. J Neuroimmunol. 2008;201-202:21-27. doi:10.1016/j.jneuroim.2008.05.020
32. Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized Trial of Thymectomy in Myasthenia Gravis [published correction appears in N Engl J Med. 2017;376(21):2097]. N Engl J Med. 2016;375(6):511-522. doi:10.1056/NEJMoa1602489
33. Guillermo GR, Téllez-Zenteno JF, Weder-Cisneros N, et al. Response of thymectomy: clinical and pathological characteristics among seronegative and seropositive myasthenia gravis patients. Acta Neurol Scand. 2004;109(3):217-221. doi:10.1034/j.1600-0404.2003.00209.x
34. Meyer DM, Herbert MA, Sobhani NC, et al. Comparative clinical outcomes of thymectomy for myasthenia gravis performed by extended transsternal and minimally invasive approaches. Ann Thorac Surg. 2009;87(2):385-391. doi:10.1016/j.athoracsur.2008.11.040
35. Bachmann K, Burkhardt D, Schreiter I, et al. Long-term outcome and quality of life after open and thoracoscopic thymectomy for myasthenia gravis: analysis of 131 patients. Surg Endosc. 2008;22(11):2470-2477. doi:10.1007/s00464-008-9794-2
36. Dhar R. Neuromuscular respiratory failure. Critical Care Neurol. 2009;15(3):40-67. doi:10.1212/01.CON.0000348809.76308.c0
37. Kukulka K, Gummi RR, Govindarajan R. A telephonic single breath count test for screening of exacerbations of myasthenia gravis: a pilot study. Muscle Nerve. 2020;62(2):258-261. doi:10.1002/mus.26987
38. Wu JY, Kuo PH, Fan PC, Wu HD, Shih FY, Yang PC. The role of non-invasive ventilation and factors predicting extubation outcome in myasthenic crisis. Neurocrit Care. 2009;10(1):35-42. doi:10.1007/s12028-008-9139-y
39. UCB presents efficacy and safety results for zilucoplan and rozanolixizumab in generalized myasthenia gravis. ucb.com/stories-media/Press-Releases/article/UCB-presents-efficacy-and-safety-results-for-zilucoplan-and-rozanolixizumab-in-generalized-myasthenia-gravis. Accessed January 11, 2023.
Dr. Morren and Dr. Li are both staff neurologists in the Neuromuscular Center in Cleveland Clinic’s Neurological Institute.
Advertisement

An overview of associated antibodies, therapies for antibody-positive disease and the outlook for atypical forms of MG

Retrospective analysis assesses frequency and severity relative to a reference antibiotic

Who is at risk? What tests should be ordered?

Guidance for the 5% to 20% of patients with suboptimal response or tolerability

Adequate dosing may improve outcomes in well-selected patients, large Cleveland Clinic series suggests

Reimagining the outpatient neurological visit with routine capture of neuroperformance data

Free portal helps researchers classify and share data using the IC-CoDE framework

Large study shows rural patients are less apt to be discharged to inpatient rehab, hampering outcomes