Locations:

Guidance for the 5% to 20% of patients with suboptimal response or tolerability

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/87a111f1-9ca2-4a20-9032-01538840f389/Pharmacy-1010456164)
Pharmacist handing medications to patient
By Christopher Zust, MD, and John A. Morren, MD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Note: This article is reprinted from the Cleveland Clinic Journal of Medicine(2023;90[2]:81-84).
If a patient with myasthenia gravis (MG) has been taking adequate doses of a first-line medication, typically pyridostigmine, for a sufficient duration but without significant efficacy, or has experienced substantial adverse effects, it may be time to consider immunosuppressive therapy. In 5% to 20% of patients, there may be suboptimal efficacy or prohibitive adverse effects with high-dose corticosteroid therapy over a period of a few weeks to three months.1-3 For these patients, nonsteroidal immunosuppressive therapy should be considered early instead of continuing high-dose corticosteroids for a longer duration. A targeted examination will help determine whether pyridostigmine or other treatment has failed.
MG, the most common disorder of neuromuscular junction transmission, results from an antibody-mediated attack against postsynaptic components of the neuromuscular junction. Clinical manifestations are typically categorized into generalized MG (GMG) or ocular MG subtypes.
Acetylcholine receptor (AChR) antibodies are the most common, and the condition is referred to as AChR antibody-positive MG (AChR+ MG). Other antibodies have been identified, including those against muscle-specific tyrosine kinase (MuSK) and lipoprotein receptor-related protein 4 (LRP4). Antibody seronegativity occurs in fewer than 10% of GMG patients and fewer than 50% of patients with ocular MG.4 Treatment recommendations are similar for seropositive AChR, seropositive LRP4 and seronegative GMG, while there are important clinical differences and specific treatment considerations with MuSK antibody-positive MG. This discussion focuses on AChR+ MG, which accounts for about 85% of cases of GMG.4
Advertisement
First-line pharmacologic management of AChR+ MG is symptomatic treatment with acetylcholinesterase inhibitors, and pyridostigmine is the only agent used routinely in the clinical setting. Corticosteroids are also used, mainly in patients with marked weakness or poor response to pyridostigmine.
When pyridostigmine doses exceed 120 mg every 3 hours, or a total daily dose of 960 mg, adverse effects (including risk of cholinergic crisis) tend to outweigh benefits. However, if a patient needs more than 240 mg of pyridostigmine per day, it is usually beneficial to move on to immunotherapy. Patients with limited symptoms, such as mild ptosis and facial weakness, who respond well to pyridostigmine may not need immunosuppressive agents or thymectomy4 (considered a first-line therapeutic option for certain patients with AChR+ MG without thymoma).5
Complete stable remission in MG is typically defined as one year with no signs or symptoms of MG without therapy, although some isolated weakness of eyelid closure is generally considered acceptable. Given the difficulty of achieving complete stable remission, an international consensus panel has proposed minimal manifestation status or better with only mild adverse effects as a reasonable therapeutic goal.5 Minimal manifestation status is characterized as no functional limitations but some muscle weakness on examination.
Corticosteroids have been shown in several studies, including two controlled trials, to be effective in MG treatment.4 The typical starting daily dose for prednisone is 20 to 60 mg, with the lower-range doses used for patients with mild to moderate symptoms. Depending on the treatment response and tolerance profile, the dose may be increased by 10 mg per day every one to two weeks, up to about 60 mg daily. An alternative high-dose regimen consists of prednisone 1.0 to 1.5 mg/kg/day, but usually not exceeding 100 mg/day.6
Advertisement
Given the many potential adverse effects of long-term corticosteroid therapy, prednisone is carefully tapered to the lowest effective dose that maintains therapeutic benefit. Weaning should be started when MG symptoms have significantly improved but need not wait until maximal efficacy is reached. In most patients, improvement is achieved after several weeks of higher-dose corticosteroids. Weaning starts with a slow taper (i.e., a dose reduction of 5 to 10 mg per month) until a daily dose of less than 5 mg is reached. At that point, a very slow taper of 1 mg per month may help to avoid relapse.7
The preferred goal of minimal but effective prednisone dosing is less than 7.5 mg daily, as this avoids most adverse effects of long-term corticosteroid use.8,9 When a patient needs maintenance prednisone dosing greater than 7.5 mg daily, other nonsteroidal immunosuppressive therapies should be considered. Steroid “dipping” is a well-described phenomenon of paradoxical worsening of symptoms in some MG patients, with manifestations that range from mild symptoms to, less commonly, respiratory failure. Accordingly, close monitoring is essential when patients start corticosteroids, especially high-dose regimens.
If the patient has been taking adequate doses of first-line medications (Table 1)5,6 for a sufficient duration but without significant efficacy or has experienced substantial adverse effects, it may be time to consider nonsteroidal immunosuppressive therapy. A targeted examination will help determine if treatment has failed. For example, worsening fatigable weakness in the limbs and the craniobulbar and respiratory muscles despite standard treatment is an indicator that pyridostigmine and corticosteroids are not controlling MG deficits. Fatigable weakness in MG typically has a diurnal pattern, worsening in the evening. In general, weakness predominates in proximal muscles, which may mimic a myopathy, but weakness is typically exacerbated with repetitive or sustained action and improves with rest.
Advertisement
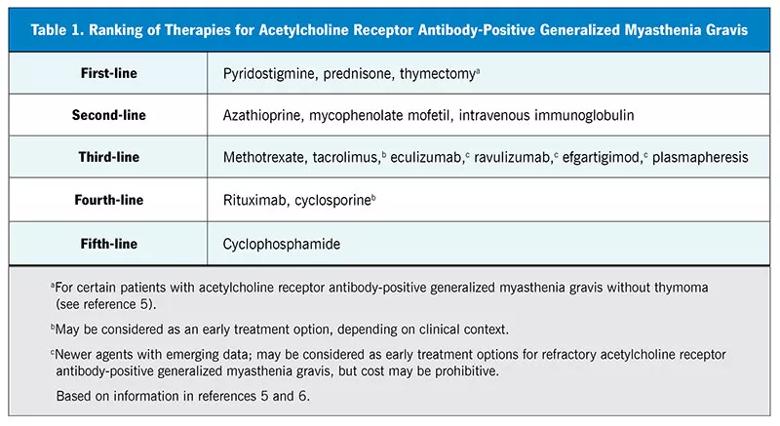
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/6ed28d08-9834-4ca0-a446-3fcc4b363c6d/23-NEU-3863416-CQD-Inset-Table1-v2-800x437-1_jpg)
Significant breakthrough symptoms warrant prompt follow-up to look for corroborating signs of uncontrolled MG. In addition to the standard examination, other key components should be included (Table 2) to rule out an impending myasthenic crisis — a life-threatening exacerbation that may necessitate intubation or mechanical ventilation. Components of the examination include the following:
Notably, pulse oximetry is not a reliable indicator of impending neuromuscular respiratory failure. The problem is not diffusion abnormality across respiratory membranes but rather carbon dioxide retention due to impaired ventilation. Generally, oxygen saturation drops only when neuromuscular respiratory impairment is well advanced.
Advertisement
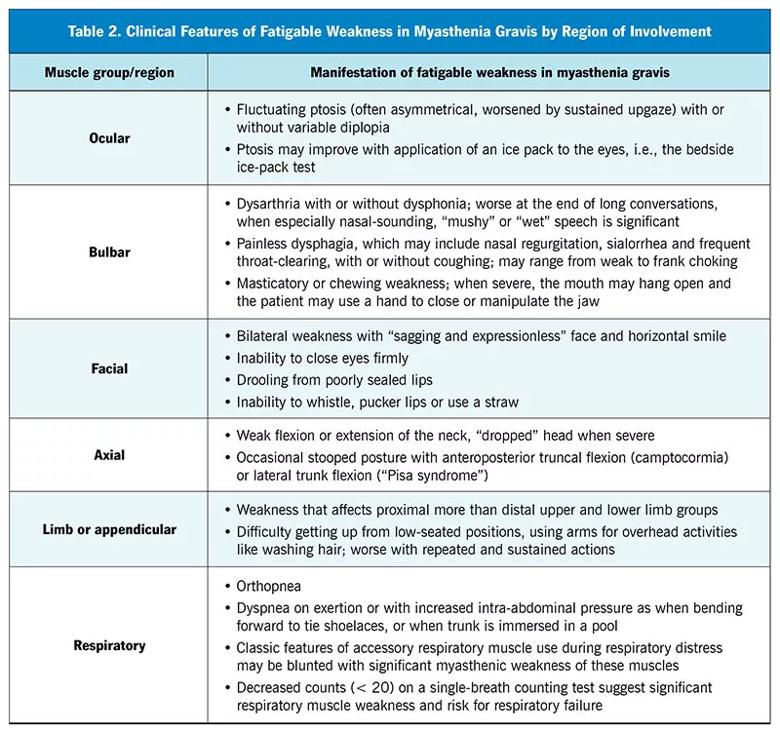
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/4a91db0d-4f3e-4000-9462-628ebbb60ee3/23-NEU-3863416-CQD-Inset-Table2-v2-800x752-1_jpg)
A patient who has features of impending myasthenic crisis such as breathing abnormalities requires prompt admission, possibly to the intensive care unit. Rescue treatments, including plasma exchange or intravenous immunoglobulin, will likely be needed.
Before appropriate treatment can be started, it is necessary to determine whether breathing abnormalities are due to MG or to another cause such as underlying heart failure, chronic obstructive pulmonary disease, asthma or pneumonia. Breathing impairment in MG typically manifests with prominent orthopnea (i.e., difficulty breathing when lying flat). Breathing difficulty may be exacerbated when the trunk is immersed, as in a pool, or when the patient bends over as when tying shoelaces, because the weakened diaphragm is unable to counteract upwardly displaced abdominal contents. Dyspnea on exertion that is disproportionate to other symptoms, lower-extremity swelling, venous distention and adventitious breath sounds (e.g., rales, wheezing and rhonchi) on auscultation can point to other non-MG causes.
Response to corticosteroid therapy in MG is classified as good or poor. A good response is characterized by a smooth response to moderate-dose (about 10 to 30 mg prednisone daily) or high-dose (about 40 to 60 mg or more prednisone daily) therapy, with remission maintained after tapering to low-dose prednisone without the need for nonsteroidal immunosuppressive therapy.
For the 5% to 20% of MG patients with a poor response after several weeks to three months of high-dose therapy,1-3 rather than continue high-dose corticosteroids, nonsteroidal immunosuppressive therapy should be considered. This therapy is often started before or at the start of steroid weaning. Current agents generally allow for long-term adequate MG control, often minimize the need for pyridostigmine, and spare the patient the adverse effects of high-dose or long-term corticosteroid therapy. For MG patients who are refractory to treatment or require more complex treatment strategies (beyond first-line agents), early input from a neurologist specializing in neuromuscular medicine and with MG expertise is highly recommended.
Immunosuppressive therapy in MG is usually associated with decreased pathologic antibody levels, but there are no evidence-based recommendations for routine measurement of these during treatment. Some data suggest that high antibody levels predict a more severe disease course.12 However, there is significant heterogeneity of clinical features, treatment response and disease course among patients with comparable antibody levels. Similarly, routine use of electrodiagnostic testing to monitor MG treatment is not well supported. Ultimately, MG disease activity is more reliably assessed clinically, so close follow-up and serial examinations are key.
1. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15(3):291-298. doi:10.1002/ana.410150316
2. Sghirlanzoni A, Peluchetti D, Mantegazza R, Fiacchino F, Cornelio F. Myasthenia gravis: prolonged treatment with steroids. Neurology. 1984;34(2):170-174. doi:10.1212/wnl.34.2.170
3. Cosi V, Citterio A, Lombardi M, Piccolo G, Romani A, Erbetta A. Effectiveness of steroid treatment in myasthenia gravis: a retrospective study. Acta Neurol Scand. 1991;84(1):33-39. doi:10.1111/j.1600-0404.1991.tb04899.x
4. Morren J, Li Y. Maintenance immunosuppression in myasthenia gravis, an update. J Neurol Sci. 2020;410:116648. doi:10.1016/j.jns.2019.116648
5. Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021;96(3):114-122. doi:10.1212/WNL.0000000000011124
6. Farmakidis C, Pasnoor M, Dimachkie MM, Barohn RJ. Treatment of myasthenia gravis. Neurol Clin. 2018; 36(2):311-337. doi:10.1016/j.ncl.2018.01.011
7. Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve. 2004;29(4):484-505. doi:10.1002/mus.20030
8. Curtis JR, Westfall AO, Allison J, et al. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis Rheum. 2006;55(3):420-426. doi:10.1002/art.21984
9. Huscher D, Thiele K, Gromnica-Ihle E, et al. Dose-related patterns of glucocorticoid-induced side effects. Ann Rheum Dis. 2009;68(7):1119-1124. doi:10.1136/ard.2008.092163
10. UK Research and Innovation. Medical Research Council: aids to the examination of the peripheral nervous system. Updated January 15, 2016. https://www.ukri.org/publications/aids-to-the-examination-of-the-peripheral-nervous-system/. Accessed January 11, 2023.
11. Juel VC. Myasthenia gravis: management of myasthenic crisis and perioperative care. Semin Neurol. 2004; 24(1):75-81. doi:10.1055/s-2004-829595
12. Heldal AT, Eide GE, Romi F, Owe JF, Gilhus NE. Repeated acetylcholine receptor antibody-concentrations and association to clinical myasthenia gravis development. PLoS One. 2014; 9(12):e114060. Published 2014 Dec 2. doi:10.1371/journal.pone.0114060
Dr. Zust, a former Cleveland Clinic neuromuscular fellow, is now a staff neurologist with McLeod Neurological Associates, Florence, South Carolina. Dr. Morren is a staff neurologist with the Neuromuscular Center in Cleveland Clinic’s Neurological Institute.
Advertisement

An overview of associated antibodies, therapies for antibody-positive disease and the outlook for atypical forms of MG

Retrospective analysis assesses frequency and severity relative to a reference antibiotic
Guidance on instructing patients, treatment selection, impact of disease course and more

Who is at risk? What tests should be ordered?

Findings may have implications for understanding the disorders’ pathophysiology

Routine capture of standardized neuroperformance data may expand and refine investigations

Nearly one-fifth of such cases fulfill 2024 McDonald criteria based on biomarkers

Adequate dosing may improve outcomes in well-selected patients, large Cleveland Clinic series suggests