Locations:

An overview of PAP and our treatment approach
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Pulmonary alveolar proteinosis (PAP) is a lung disease in which the failure of alveolar macrophages to clear surfactant causes its accumulation in the alveoli, leading to impaired gas exchange and hypoxemia. PAP is rare, more common in men and typically presents between 30 and 60 years of age.
The clinical course is variable, ranging from spontaneous resolution to death caused by progressive respiratory failure or infections. My colleagues and I recently reviewed the pathophysiology of and clinical approach to PAP in The Lancet Respiratory Medicine, summarized below.
The underlying pathophysiology of all forms of adult PAP is abnormal alveolar macrophage maturation and surfactant clearance. Elucidation of the role of granulocyte-macrophage colony-stimulating factor (GM-CSF) in mediating macrophage maturation and surfactant catabolism greatly advanced our understanding of PAP.
In autoimmune PAP, anti-GM-CSF antibodies lead to impaired bioavailability of GM-CSF, which disrupts the GM-CSF signaling pathways that regulate alveolar macrophage maturation and function. Autoimmune PAP is the most common form of the disease and accounts for approximately 90 percent of cases of adult PAP.
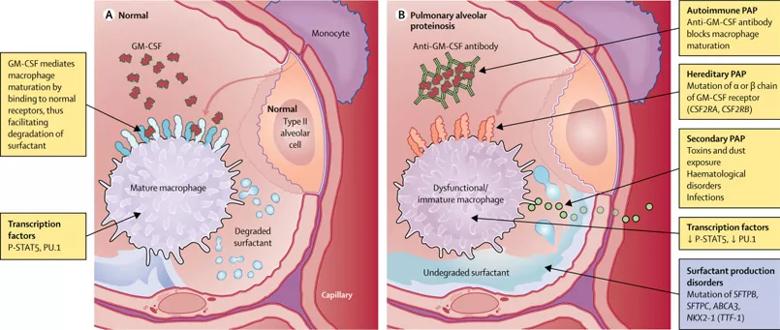
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/159cc7c2-740a-490b-95b1-7f0d2bcddf54/18-PUL-294-Culver-Inset-Image-805pxl-width_03_jpg)
Image republished with permission from Elsevier.
A number of hematological and environmental factors, including hematological malignancies, primary immunodeficiency diseases, inhalational exposures (dust), infections and drugs, cause abnormal alveolar macrophage function that leads to secondary PAP, characterized by the lack of anti-GM-CSF antibodies. Congenital (or hereditary) PAP is attributed to genetic mutations in GM-CSF receptor proteins or surfactant proteins.
Advertisement
Clinical findings and pulmonary function tests
Symptoms of autoimmune PAP include dyspnea, chest discomfort and constitutional symptoms. Cough is a common symptom, but up to a third of patients may be asymptomatic. Physical examination is usually nonspecific, although inspiratory crackles and clubbing may be observed.
Pulmonary function tests usually indicate restrictive impairment and reduced single-breath diffusing capacity of carbon monoxide (DlCO). Arterial blood gas measurements can reveal hypoxemia and elevation of the alveolar-arterial oxygen gradient.
Imaging studies
Chest radiographs typically show bilateral symmetric alveolar opacities located centrally in the mid and lower lung zones, often in a “bat wing” distribution. Distinctive features on high-resolution computed tomography (HRCT) include patchy ground glass opacities and smooth thickening of intralobular septal lines. The mosaic pattern of polygonal shapes composed of ground glass outlined by septal thickening has been referred to as “crazy paving” and is highly characteristic, albeit not pathognomonic, of PAP.
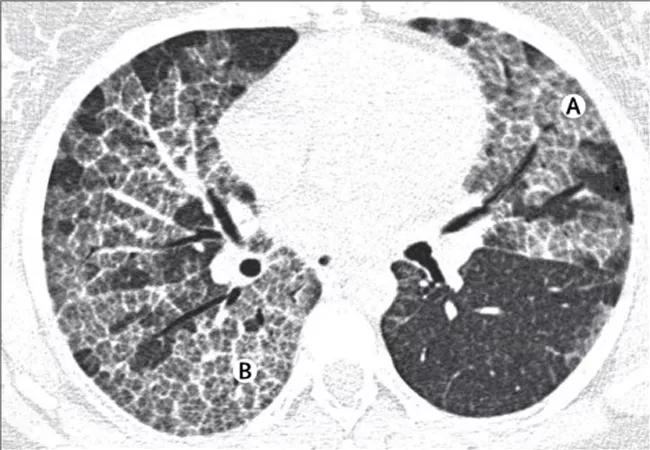
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/45f62de3-4a12-43ca-b99f-6d7f87e5dfd2/18-PUL-294-Culver-Inset-Image-650pxl-width_02_jpg)
HRCT images show diffuse ground-glass opacification (A) and the “crazy paving” pattern (B). Republished with permission from Elsevier.
Bronchoscopy
Bronchoscopy can confirm a diagnosis of PAP in most patients. Bronchoalveolar lavage (BAL) usually yields opalescent or milky-appearing fluid due to the high lipoproteinaceous content of the amorphous material in the alveolar spaces (Figure 3). Transbronchial lung biopsy (TBLB) can be considered if the BAL is not characteristic of PAP. The high yield of TBLB eliminates the need for a surgical lung biopsy in most patients; surgical biopsy to confirm PAP is required in only 10-20 percent of cases.
Advertisement

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/392599e5-c914-4aff-8076-dca6fcd4512c/18-PUL-294-Culver-Hero-Image-650-pxl-width_01_jpg)
Characteristic milky or opalescent appearance of fluid return from whole-lung lavage in a patient with PAP. L1 and R1 show the drained fluid after the first lavage cycle in the respective left and right lung; L2 and R2 bottles were collected after 9 L of lavage; and L3 and R3 show the clear drained fluid at the last lavage cycle, in this case after a total of 30 L per lung. Republished with permission from Elsevier.
Serologic tests and biomarkers
Antibodies to GM-CSF in serum and BAL fluid are detected in nearly all patients with autoimmune PAP but not in patients with secondary or congenital PAP. Although the presence of anti-GM-CSF antibodies is an extremely useful diagnostic test, the serum concentration of GM-CSF antibodies does not appear to correlate with severity of autoimmune PAP.
Elevated serum concentrations of lactic dehydrogenase (LDH) have been reported in up to 80 percent of patients with PAP. High concentration of LDH has been shown to correlate directly with an alveolar-arterial gradient and inversely with partial pressure of arterial oxygen (PaO2) but is considered nonspecific.
Many other biomarkers have been investigated, but none are widely accepted in clinical practice to determine severity of PAP or to monitor disease progression. Current standards in the U.S. include extent of imaging findings, degree of gas exchange impairment (e.g., A-a gradient, six-minute walk test, oximetry), other pulmonary function tests and patient symptoms.
Patients who are asymptomatic and who do not have substantial physiological impairment (e.g., hypoxia) can be monitored by pulmonary function tests, symptom assessments and chest radiography. Intervention is warranted for patients with moderate-to-severe PAP. Patients with secondary PAP should be treated for the underlying conditions.
Therapeutic whole-lung lavage
Therapeutic whole-lung lavage (WLL) has been the cornerstone of treatment of PAP for decades, and approximately 80 percent of patients experience improvement with an initial implementation of WLL. Our Interstitial Lung Disease Program at Cleveland Clinic has long served as a referral center for PAP and was among the first to develop same-day bilateral WLL.
Advertisement
Patients are usually able to return to their homes the day after the procedure. WLL has dramatically improved the natural history of PAP and continues to be the standard initial treatment in patients with clinically significant lung disease.
Therapeutic bronchoalveolar lavage (BAL)
Segmental lobar lavage performed with fiberoptic bronchoscopy, while inferior to WLL, may be reserved for patients who are unable to undergo WLL under general anesthesia.
Additional treatment modalities
Other therapeutic approaches to PAP that have been less commonly employed include subcutaneous and aerosolized GM-CSF and rituximab infusions. Subcutaneous and inhaled recombinant GM-CSF are being investigated as initial treatment or as a sequel to WLL for patients with autoimmune PAP; however, large-scale studies directly comparing the efficacy of GM-CSF therapy to WLL are currently lacking.
Rituximab, a monoclonal antibody directed against the CD20 antigen, has shown promise in treatment of autoimmune PAP, though experience with this treatment is limited. Additional studies are needed to determine the efficacy of rituximab as well the patients most likely to benefit from therapy.
Given that WLL is effective for a large proportion of patients, the use of these alternative therapies must be carefully considered and tailored to each individual. Lung transplantation is a potential consideration for patients with severe, refractory PAP, but underlying genetic PAP should be ruled out based on concern for a recurrence in the lung allograft.
Advertisement
The management of PAP at Cleveland Clinic is collaborative, individualized and innovative. Our bronchoscopy team and thoracic surgeons are able to perform both standard and advanced procedures when a biopsy is required for diagnosis. The extensive experience of our team helps us to avoid invasive diagnostic procedures when possible, and we continue to pursue advances in the diagnosis and treatment of this rare disease.
Dr. Culver directs the Interstitial Lung Disease Program in the Department of Pulmonary Medicine at Cleveland Clinic.
Advertisement

Recent breakthroughs have brought attention to a previously overlooked condition

A review of treatment options for patients who may not qualify for surgery

Looking at the real-world impact and the future pipeline of targeted therapies

The progressive training program aims to help clinicians improve patient care

New breakthroughs are shaping the future of COPD management and offering hope for challenging cases

Exploring the impact of chronic cough from daily life to innovative medical solutions

How Cleveland Clinic transformed a single ultrasound machine into a cutting-edge, hospital-wide POCUS program

Collaborative patient care, advanced imaging techniques support safer immunotherapy management