Locations:

A Cleveland Clinic pulmonologist highlights several factors to be aware of when treating patients
Written by Kamonpun Ussavarungsi, MD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Diffuse cystic lung diseases encompass a complex and rare condition that is frequently characterized by a convergence of causes. These factors may encompass elements such as genetic predisposition, exposure to environmental agents like tobacco, autoimmune responses and/or neoplastic processes. Navigating the distinction between labeling a condition as a diffuse disease or opting for an aggressive investigation poses a challenge. Several diseases can be associated with pulmonary cysts, so it’s important to consider each case individually to determine what the presence of cysts can mean in terms of treatment.
Age can play a role in the incidence rate of pulmonary cysts, and this is a factor that must be considered when determining treatment options. For example, in the Framingham Heart Study offspring and third-generation cohort comprising 2,633 individuals (with a mean age of 59.2 years), the prevalence of pulmonary cysts is noted at 7.6%, and this prevalence tends to rise with age.1 It is also plausible that cystic lesions may be intrinsic to the aging process. Specifically, the discovery of fewer than five cysts, particularly when isolated and observed in an elderly patient without a notable medical history, may be considered incidental.
Nevertheless, the manifestation of pulmonary cysts in a young population may suggest an association with specific conditions. Rare hereditary disorders linked to lung cysts comprise genetic connective tissue disorders like Marfan’s syndrome and Ehlers–Danlos syndrome. Additionally, conditions such as COPA syndrome, arising from a mutation in the COPA gene, involve a combination of genetic factors and autoimmune or autoinflammatory responses, with lung cysts forming part of their clinical manifestations. Indeed, certain syndromes may present with pneumothorax and display authentic cysts, hyperlucency, or emphysema-like lesions on CT scans. Examples include Birt-Hogg-Dubé syndrome, linked to an FLCN mutation, and Proteus syndrome, associated with an AKT1 mutation. In such cases, the coexistence of pneumothorax and distinctive extrapulmonary imaging findings can indicate the presence of these specific genetic disorders.
Advertisement
Certain disease groups exhibit higher awareness and understanding, as exemplified by lymphangioleiomyomatosis (LAM). This disease presents with distinctive characteristics and patterns in the distribution of cysts, offering suggestive clues for specific conditions. LAM is classified into sporadic cases and those related to tuberous sclerosis complex (TSC).
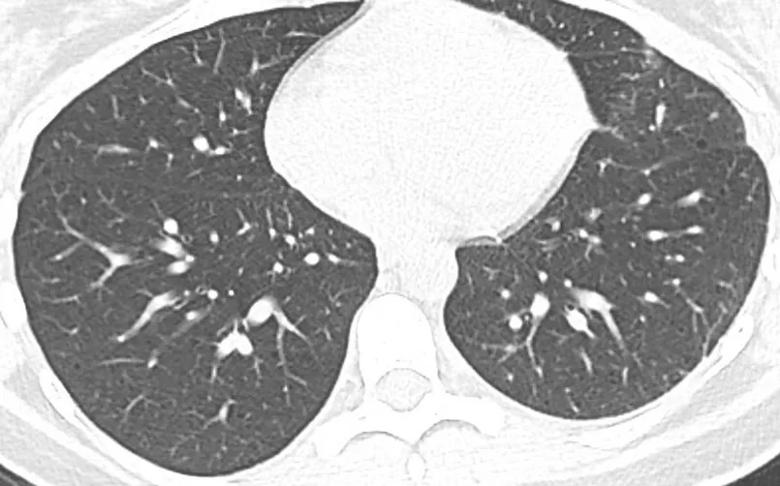
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/dfc4df68-449e-4714-93d6-d69cf5026364/Diffused-Cystic-Lung-Disease-1_jpg)
Figure 1. A female patient with a history of recurrent pneumothoraces and a subtle, easily overlooked cyst, received a diagnosis of Lymphangioleiomyomatosis (LAM) through a surgical lung biopsy performed concurrently with pleurodesis.
Real-life medical practice often presents challenges, and CT imaging may reveal ambiguous or atypical findings that differ from textbook depictions. From our experience, in the early stages of LAM, tiny cysts can be easily overlooked (Figure 1). Conversely, advanced cases may sometimes resemble other advanced-stage cystic damage, making differentiation challenging (Figure 2).
In some patients, appropriate extrapulmonary findings, such as kidney angiomyolipoma, lymphangioleiomyomas or elevated vascular endothelial growth factor-D (VEGF-D) levels, are sufficient for making a LAM diagnosis without the need for a biopsy. The VEGF-D test is a specialized examination requiring a specific form for sending out, and it is important to note that the routinely ordered VEGF test, commonly dispatched, differs from VEGF-D.
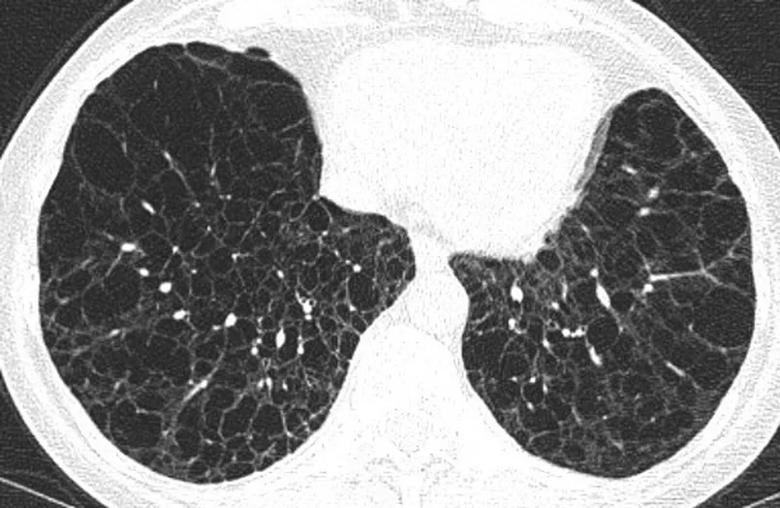
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/0d54410c-de32-4869-8492-3b75cc2639fe/Diffused-Cystic-Lung-Disease-2_jpg)
Figure 2. A female patient in an advanced stage of cystic lung disease was diagnosed with Lymphangioleiomyomatosis (LAM), supported by evidence of extrapulmonary features, including AML (angiomyolipomas), chylous effusion, and an exceptionally elevated VEGF-D level exceeding 3000.
Advertisement
Some scenarios involve a combination of conditions like lymphocytic interstitial pneumonia (LIP) and amyloidoma, which may occur in association with Sjogren’s syndrome. Alternatively, pulmonary amyloidosis on a CT scan can mimic advanced smoking-related damage. (Figure 3). Clinical scenarios can be complex, with variations in presentation not uncommon. This emphasizes the importance of a comprehensive approach, considering clinical history, additional diagnostic tests and consultation with specialists to arrive at a more accurate and nuanced diagnosis.
It’s important to note that some patients with cysts maintain stability throughout their lives, experiencing minimal or no pulmonary impairment. In such cases, invasive workup may not be necessary. Each patient is unique, emphasizing the need for thorough evaluations tailored to this specific group of diseases.
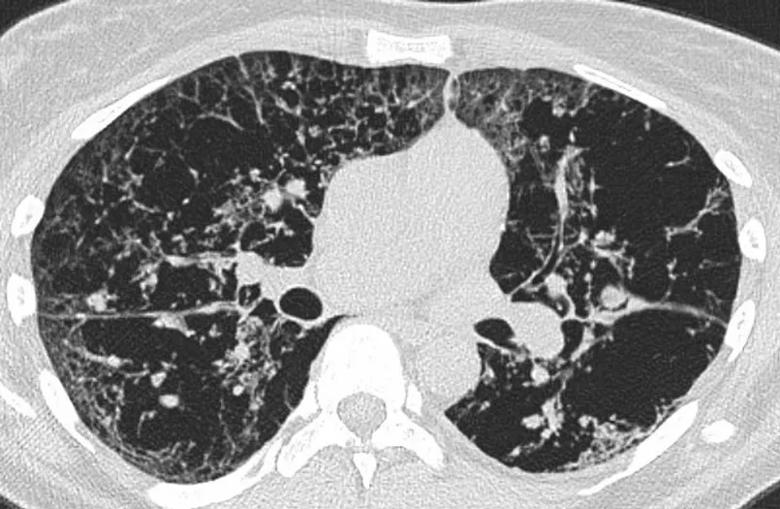
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/96ba2b74-c506-4d6f-8964-be0e72410686/Diffused-Cystic-Lung-Disease-3_jpg)
Figure 3. A female patient exhibiting extensive cystic abnormalities, presenting challenges in characterization, along with peribronchial and alveolar consolidations, has been diagnosed with light chain amyloidosis through a surgical biopsy.
Treatment approaches can vary, including the use of an mTOR inhibitor for LAM, a chemotherapy regimen for amyloidosis, surgical resection for progressive cystic lesions with pressure effects in Proteus syndrome, and immunosuppressive therapy in cases of impaired LIP.
While some conditions may lack proven therapies, lung function often maintains stability, as observed in cases like Birt-Hogg-Dubé. Often, discussions arise about tissue sampling. Based on this information, the decision to proceed with a surgical biopsy varies from patient to patient, depending on factors such as the impairment or progression of the condition, determining treatment options post-pathology results, and how the patient’s underlying condition affects their tolerance for the procedure. The less aggressive option would be a transbronchial biopsy or cryobiopsy for obtaining larger samples, with the acknowledgment of imperfections since the culprit can still be missed. A thorough discussion of the risks and benefits is crucial before proceeding.
Advertisement
At Cleveland Clinic, patients with cystic lung lesions undergo clinical evaluation by experienced pulmonologists and radiologists to assess CT findings. Subsequent investigations outlined by the pulmonologist based on potential etiology may include genetic evaluation, counseling or consultations with other specialists if extrapulmonary conditions are discovered. We always ensure that patients undergo the necessary procedure if lung tissue is needed due to unremarkable non-invasive investigations. Our pulmonary intervention team is also available, and we offer a less invasive biopsy option — transbronchial cryobiopsy — which may be a first option for some patients before turning to VATS.
Through support from the TSC Alliance and LAM Foundation, our TSC & LAM Clinic functions as a specialized center to help manage these complex conditions. We closely collaborate with other specialists to address extrapulmonary symptoms, especially in cases involving genetic syndromes, to ensure comprehensive patient care. The development of the TSC & LAM Clinic has helped our team tremendously as we handle rare cases, conduct investigations and run genetic panels. These panels have led to targeted therapy in many instances, such as somatic TSC2 mosaicism-related arm and upper torso overgrowth, where a genetic panel is run on a skin biopsy. Given the rarity of the condition, we actively participate in ongoing research and collaborate with other LAM centers through multi-center discussions.
References
Advertisement
Advertisement

Overexposure to the common alloy can lead to an autoimmune response called sensitization

A look into recent developments in the diagnosis of LAM and its clinical presentation

A review of treatment options for patients who may not qualify for surgery

Rising rates in young miners illustrate the need for consistent prevention messaging from employers and clinicians

Multidisciplinary focus on an often underdiagnosed and ineffectively treated pulmonary disease

Management and diagnostic insights from an infectious disease specialist and a pulmonary specialist

Treatments can be effective, but timely diagnosis is key
Patient experience improves with a multidisciplinary approach