Locations:

Pheochromocytoma case underscores the value in considering atypical presentations
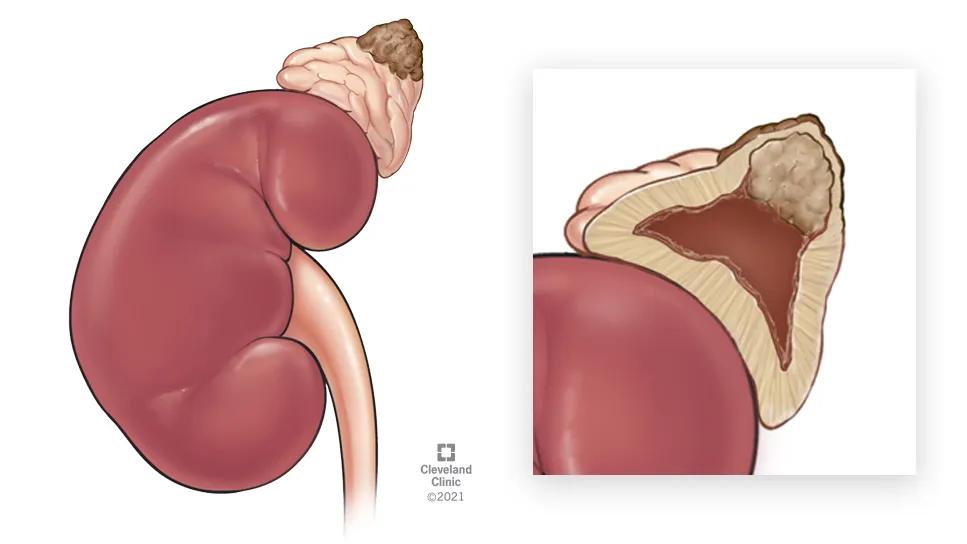
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/e3845cae-90ac-4cef-a25f-ca9b052581f6/pheochromocytoma-case-study-hero)
medical illustration of a pheochromocytoma
A recent case involving a patient cared for at Cleveland Clinic’s Endocrinology & Metabolism Institute provides a reminder to be aware that well-established diagnostic procedures can sometimes yield atypical results.
A CT scan revealed bilateral adrenal nodules in a 59-year-old woman with abdominal pain. The patient was referred to Cleveland Clinic endocrinologist Snigdha Reddy Bendaram, MD.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
“Whenever we encounter nodules, two questions come to mind,” says Dr. Bendaram. “First, are these functional nodules? Adrenal glands produce a number of hormones, and adrenal nodules can produce any of them or they can be inert.
“The second question is do we see a risk of cancer?” she adds. “The nodules might be benign or malignant, or they might be pheochromocytomas, which have a capacity to spread.”
Standard lab work in the context of a possible pheochromocytomas calls for a blood test to measure plasma-free metanephrines and normetanephrines. Presence of pheochromocytoma is typically indicated if plasma-free metanephrine levels are two to four times above normal.
The patient’s metanephrine levels were at mildly elevated but acceptable levels. “Slight elevations can be caused by medication interferences or even anxiety,” Dr. Bendaram says. “They can be commonly seen, for example, in people going in for an exam or an interview.”

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/78e99e35-999a-48a8-a094-41c702d8dda4/pheochromocytoma-case-study-inset-2)
While the patient’s hormone levels were not alarming, the size and indeterminate nature of a large nodule on the right-side adrenal gland raised concerns about malignancy. Dr. Bendaram referred the patient to endocrine surgeon Gustavo Romero Velez, MD, for right-side adrenalectomy.
Laparoscopic en-bloc resection of the gland (10.5 cm x 6.5 cm x 3.4 cm) was done to assure negative margins. The pheochromocytoma measured 5.2 cm x 4.2 cm x 3.5 cm.

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/65b961ea-ae6f-46f4-b7e0-5eb7e1d300a7/pheochromocytoma-case-study-inset-1)
Image of the patient's resected tumor.
While open surgery may be considered in cases of a large mass or suspected cancer, Dr. Romero Velez notes that the team at Cleveland Clinic’s high-volume endocrine surgery department assessed the adrenalectomy could safely be done with a minimally invasive technique that optimized the patient’s recovery.
Advertisement
Pathology findings on the lesion proved unexpected. Despite unremarkable hormone levels before surgery, the tumor was a pheochromocytoma with components of a peripheral neuroblastic tumor.
Pheochromocytomas are uncommon tumors that arise from chromaffin cells in one or, less commonly, both adrenal medullae.
An estimated 10% to 15% of pheochromocytomas are malignant, and they are categorized as localized (as this patient’s was), regional, metastatic or recurrent. Common symptoms typically relate to excessive hormone production — hypertension, headache, diaphoresis and arrhythmias.
Our patient’s tumor had histologic features associated with aggressive behavior: invasion into periadrenal adipose tissue, diffuse/large nested growth, tumor spindling and profound nuclear pleomorphism. Her PASS score (pheochromocytoma of the adrenal gland scaled score) was 7; a score of 4 or more indicates higher risk for aggressive behavior.
Genetic conditions linked to pheochromocytomas are found in 25% to 35% of people with the tumors. Those conditions include multiple endocrine neoplasia 2 types A and B (MEN2A and MEN2B), von Hippel-Lindau disease, neurofibromatosis type 1, hereditary paraganglioma syndrome, Carney-Stratakis dyad and the Carney triad. Genetic testing may be appropriate in cases of multiple pheochromocytomas, diagnosis of pheochromocytoma before age 40, or a family history suggesting a predisposition.
Surgery is the primary treatment for pheochromocytomas and is successful in 90% of cases.
Advertisement
While pheochromocytomas typically cause excess hormone production, it isn’t always true.
“The first thing that I did was went back to make sure that I didn't miss something, but no, nothing was elevated,” says Dr. Romero Velez. “I showed the results to my partner, who has been in practice for 30 years or so. In rare cases, he said, you can come across cases where the tumor is not producing hormones.”
In patients with pheochromocytoma diagnosed pre-operatively, surgical preparation starts with using alpha adrenergic receptor blockers followed by beta adrenergic blockers, which thwarts hypertensive emergencies that can happen during surgery. In this case, the patient had slightly elevated blood pressure toward the end of surgery, Dr. Romero Velez says, but the procedure otherwise proceeded smoothly.
Since surgery, the patient’s metanephrines, imaging and genetic testing all have been normal. The minimally invasive surgical approach supported a good recovery.
Advertisement
Advertisement

Procedure offers potential for less pain and faster recovery in selected patients

Challenges include multimorbidity, polypharmacy, limited mobility and sarcopenia

Data could help inform policy and patient-provider conversations

Advocacy group underscores need for multidisciplinary expertise

A reconcilable divorce

A review of the latest evidence about purported side effects

High-volume surgery center can make a difference

Advancements in equipment and technology drive the use of HCL therapy for pregnant women with T1D