Locations:

From early symptoms to treatment
Neurofibromatosis types 1 (NF1) and 2 (NF2) are rare genetic conditions inherited in an autosomal dominant manner that predominantly affect the skin and nervous system.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
“One of the defining characteristics of both conditions is the propensity for the development of both nonmalignant and malignant tumors,” says Neil Friedman, MBChB, Director of Cleveland Clinic’s Center for Pediatric Neurosciences. “In NF1, children are predisposed to low-grade glial cell tumors (gliomas) of the optic pathway and brainstem, whereas in NF2, tumors of the cranial nerves, especially vestibular schwannomas, meninges (meningioma) and spinal cord (ependymomas) are more typical.”
“An important aspect of managing NF1 and NF2 is treating the associated tumors,” adds pediatric neuro-oncologist Tanya Tekautz, MD, in Cleveland Clinic’s Department of Pediatric Hematology/Oncology and Blood and Bone Marrow Transplantation. “In approximately two thirds of patients, we are able to avoid the use of radiation and shrink or stabilize the size of optic gliomas using chemotherapy. Meningiomas and ependymomas associated with NF2 are typically harder to treat, because chemotherapy is not as effective, and radiation and surgery carry risks.”
Of the two types of neurofibromatosis, NF1 is more common — it affects approximately one in 3,000 individuals, while NF2 afflicts approximately one in 40,000 individuals. An estimated 50 percent of all neurofibromatosis cases are inherited from an involved parent, while 50 percent arise as spontaneous mutations.
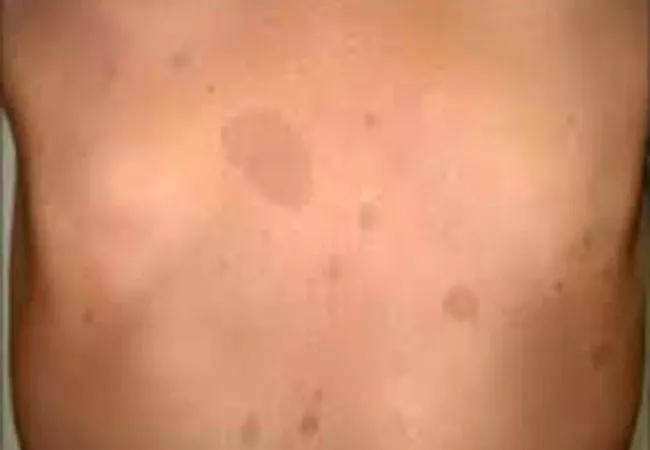
Although most children with NF1 develop defining clinical features by the age of 10, the high variability in phenotypic expression can sometimes delay diagnosis. The most common initial presenting signs include the neurocutaneous features, namely café au lait macules (CALMs), freckling and Lisch nodules.
Advertisement
“CALMs are typically the initial symptom of NF1,” explains Dr. Friedman. “Although isolated café au lait macules are not uncommon, pediatricians should be aware that multiple CALMs (> 6 mm) raise the suspicion for neurofibromatosis. In a prepubertal child, café-au-lait macules measuring >5 mm in size are considered significant for diagnostic criteria of NF1, whereas the size requirement is >15 mm in a postpubertal child.”
Freckling is another important feature of NF1.
“There is a predilection for freckling in ‘hidden areas’ such as the armpits, groin, or, in women, the folds of the breast,” notes Dr. Friedman. “Axillary or groin freckling is one of the diagnostic criteria for NF1. Lisch nodules, or benign hamartomas or tumors of the iris, another diagnostic criteria, are rarely seen before age 5 or 6 and are present in most people by their late 20s.”
According to Dr. Friedman, the hallmark of NF1, the neurofibroma or peripheral nerve sheath tumor, is the most common tumor seen in NF1 and undergoes malignant transformation in <10 percent of cases. The neurofibromas may also coalesce or occur on a cluster of nerves forming a complicated or plexiform neurofibroma.
“Diagnostic criteria for NF1 require two or more cutaneous or subcutaneous neurofibromas or one plexiform neurofibroma,” he says. “The neurofibromas may not always be visible to the eye or felt by palpation but can occur deep within the tissues or the body.”
Differentiating nonmalignant from malignant neurofibromas or neurofibrosarcomas can be difficult.
Advertisement
“Pain or an enlarging mass, especially in a pre-existing neurofibroma, should alert the pediatrician to possible malignant concern,” says Dr. Friedman. “Neurofibromas may also result in cosmetic disfigurement or functional impairment when they occur over a joint, for example.”
Dr. Tekautz explains that the treatment of malignant neurofibromas poses a significant challenge to the treating clinician.
“The growth of malignant peripheral nerve sheath tumors are historically been treated with a combination of radiation and chemotherapy – doxorubicin and Ifosfamide,” she says. “However, due to the resistant nature of these tumors, mutations frequently develop.”
The most common central nervous system (CNS) tumors associated with NF1 are optic gliomas, astrocytomas, and brainstem or cerebellar gliomas.
“Optic pathway lesions in children with NF1 often present before 5 years of age,” says Dr. Tekautz. “Because the body compensates for the loss of vision in one eye, it may be difficult to catch optic glioma early. Sometimes, the children tilt their head, cover the affected eye, or just preferentially use the healthy eye in the effort to compensate for the loss of vision in the affected eye.”
She notes that “approximately 10-15 percent of children who develop low-grade gliomas of the optic nerve and the optic pathway are at risk of developing loss of vision, which is why timely treatment is very important.”
“The treatment for optic gliomas consists of outpatient chemotherapy with two intravenous medications, most often a combination of carboplatin and vincristine for anywhere between 52 and 72 weeks,” she continues. “Oral medications given every 6 hours may also be an option, depending on the individual circumstances and family preferences. We try to avoid radiation and associated side effects such as neurocognitive and vascular problems. Surgery is also rarely, if ever, a consideration due to the placement of multiple tumors along the optic pathway.”
Advertisement
Unfortunately, therapeutic options for malignant peripheral nerve sheath tumors (MPNSTs) are limited.
“While surgical resection is one treatment option, nerve damage may occur as a consequence of the procedure because the tumors are intrinsic to the nerve,” says Dr. Friedman. “Others, especially deep-seated plexiform neurofibromas, may not be amenable to surgery.”
The lack of effective treatment options especially for plexiform neurofibromas and MPNSTs is the main driving force behind the NF1 therapeutic pipeline — there are currently over 25 experimental drugs for neurofibromatosis in different stages of development. On Feb. 15, 2018, the US Food and Drug Administration granted Orphan Drug Designation to selumetinib, an MEK 1/2 inhibitor, for the treatment of NF1 tumors.
Although CNS and peripheral nerve sheath tumors are some of the most feared complication of NF1, the condition is also associated with a range of other medical and neurobehavioral manifestations that warrant medical attention.
“Children with NF1 often have cognitive and learning disabilities and an increased incidence of autism, cardiac abnormalities and seizures,” says Dr. Friedman. “Macrocephaly and scoliosis are frequent accompaniments. Furthermore, children with NF1 are at an increased risk of vascular abnormalities of the brain such as stenoses, occlusions and aneurysms. Bony abnormalities/skeletal dysplasias and other malignancies such as nonlymphocytic leukemias and pheochromocytomas may rarely occur. The latter should be monitored for by regular blood pressure check at well child visits.”
Advertisement
Dr. Friedman further points out that NF-related disorders are chronic disorders that require long-term management and follow-up care, and that Cleveland Clinic provides continuity of care as individuals progress into adulthood.
“Our team approach provides individualized care with access to a broad range of specialists with the availability of cutting-edge technology, latest imaging tools, and multi-modality treatments should they be necessary,” he says.
The Cleveland Clinic Neurofibromatosis Center is a comprehensive, patient-focused clinic for the treatment of NF1 and NF2, recognized by the Neurofibromatosis Clinic Network (NFCN).
Advertisement

Aim is for use with clinician oversight to make screening safer and more efficient

Rapid innovation is shaping the deep brain stimulation landscape

Study shows short-term behavioral training can yield objective and subjective gains

How we’re efficiently educating patients and care partners about treatment goals, logistics, risks and benefits

An expert’s take on evolving challenges, treatments and responsibilities through early adulthood

Comorbidities and medical complexity underlie far more deaths than SUDEP does

Novel Cleveland Clinic project is fueled by a $1 million NIH grant

Tool helps patients understand when to ask for help