Locations:

Evaluation of a patient presenting with worsening swelling of the lips and legs
By Mohammad A. Sohail, MD, Peter Luong, John Sedor, MD, and Roop Kaw, MD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Editor’s note: This article was originally published in the Cleveland Clinic Journal of Medicine.
A 34-year-old female patient with a history of systemic lupus erythematosus (SLE) presented to the emergency department after several days of worsening swelling of the lips and legs along with decreased oral intake. She also reported intermittent pleuritic chest pain and exertional dyspnea for the past two months.
The patient was previously diagnosed with SLE with cutaneous manifestations including malar rash and discoid lesions of the face and scalp, recurrent pericardial and pleural effusions, diffuse arthralgias, and positive serologic markers including positive antinuclear antibody, anti-dsDNA antibody, anti-Smith antibody, and low complement levels. A previous pericardial pleural effusion resulted in normalization of complement levels with corticosteroid therapy.
Medical history was significant for hypertension, controlled with amlodipine, as well as two unexplained consecutive first-trimester miscarriages and recurrent pulmonary embolism. However, prior investigations were not consistent with antiphospholipid antibody syndrome, with the absence of lupus anticoagulant, anticardiolipin immunoglobulin (Ig)G/ IgM, and anti-B2 glycoprotein IgG/IgM on two separate occasions six months apart. Her home medications included the anticoagulant rivaroxaban, amlodipine, prednisone, hydroxychloroquine, methotrexate, and folic acid. However, the patient had been nonadherent for the previous 4 months owing to lack of insurance coverage and instead had been trying alternative remedies including herbal tea.
Advertisement
The patient also had lymphocytopenia, which can be associated with viral infections, active disease in SLE, and immunosuppressive therapy including glucocorticoids.
In the emergency department, the patient’s temperature was 36.7°C (98.1°F), heart rate was 92 beats per minute, blood pressure was 156/103 mm Hg, and peripheral oxygen saturation was 100% on room air.
Physical examination revealed lip-swelling, aphthous ulcers, pitting edema in both legs and feet, and a new nontender rash on palms and soles. There was no appreciable murmur, pericardial rub, or jugular venous distention. The patient was alert and oriented to time, place, and person, followed all commands, and had no focal neurologic deficits. Funduscopic examination revealed vessel attenuation, cotton wool spots, and dot-and-blot hemorrhages without any papilledema, consistent with grade III hypertensive retinopathy.
Laboratory testing revealed anemia, thrombocytopenia, and an elevated serum creatinine level of 1.82 mg/dL (increased from baseline of 0.8 mg/dL).
Urinalysis with microscopic examination yielded cloudy appearance with high hemoglobin, red blood cells, protein, granular casts, urine protein-to-creatinine ratio, and urine sediment with dysmorphic red blood cells and red blood cell casts. See Table 1 for laboratory results.
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/52553c22-5adf-4ad9-be88-d3e6e72d2428/22-URL-3019678-CQD-Inset-2-793x1024_jpg)
A chest radiograph showed significant increase of the cardiac silhouette compared with prior radiographs, and subsequent echocardiography demonstrated a large pericardial effusion.
Patient presentation of new aphthous ulcers, deteriorating kidney function, pancytopenia with lymphocytopenia, and a new large pericardial effusion was suggestive of an active lupus flare. The lip-swelling was also concerning for angioedema, which has been reported as an unusual manifestation of SLE, with the proposed mechanism being acquired deficiency of C1 esterase inhibitor.1,2
Advertisement
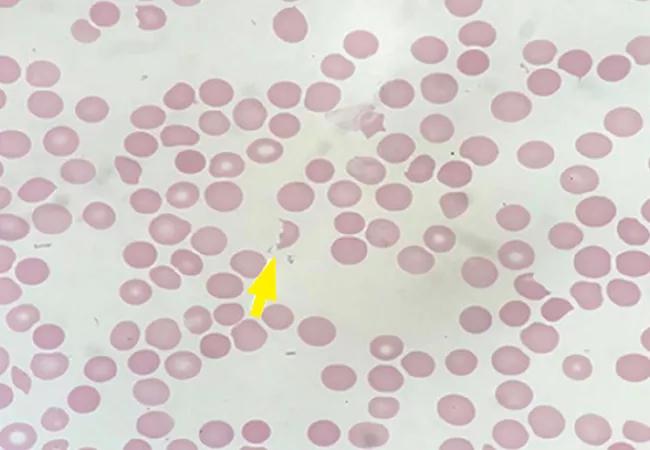
Peripheral smear revealed abundant schistocytes and thrombocytopenia (Figure 1). Other testing showed a reticulocyte index of 0.69% (indicative of hypoproliferation), a negative direct Coombs test, and prothrombin and partial thromboplastin times within normal limits.
At this point, microangiopathic hemolytic anemia (MAHA) was suspected, which is a descriptive term for nonimmune or Coombs-negative hemolysis, resulting from intravascular red blood cell fragmentation that produces schistocytes on peripheral blood smear.
Further laboratory test results included low C3, low C4, high dsDNA, positive Crithidia lucillae, negative lupus anticoagulant, negative anticardiolipin IgG and IgM, negative anti-B2 glycoprotein IgG and IgM, low ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity, and negative ADAMTS13 inhibitor.
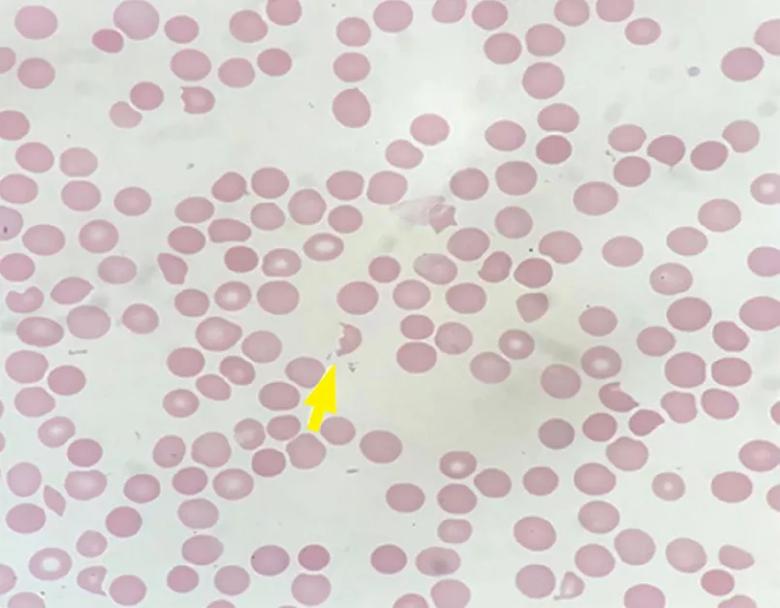
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/72c0e65e-ffb3-4349-b197-0494c8e6db44/22-URL-3019678-CQD-Inset-1_jpg)
The combination of characteristic laboratory data in our patient, including negative Coombs test, elevated lactate dehydrogenase (LDH), low haptoglobin, normal coagulation studies, and peripheral smear showing abundant schistocytes, were characteristic for MAHA. The combination with thrombocytopenia and worsening kidney function raised high suspicion for TMA.
Advertisement
TMA is a pathologic diagnosis determined by tissue biopsy in which abnormalities of arteriolar and capillary walls subsequently lead to microvascular thrombosis. As described previously, MAHA refers to Coombs-negative hemolysis resulting from intravascular red blood cell destruction, which may be caused by abnormalities in the microvasculature or even by intravascular devices such as prosthetic heart valves. Consequently, even though not all MAHA is caused by TMA, the diagnosis of TMA can be typically inferred in patients with MAHA and thrombocytopenia in the appropriate clinical setting.
TMA syndrome encompasses many disorders that include TTP, Shiga toxin-induced hemolytic uremic syndrome, drug-induced TMA, radiation-induced TMA, and complement-mediated TMA, which is also referred to as atypical hemolytic uremic syndrome.
Systemic disorders that can cause TMA include scleroderma, severe hypertension, preeclampsia/HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome, and antiphospholipid syndrome, especially the most severe form of antiphospholipid syndrome known as catastrophic antiphospholipid syndrome, which is characterized by disseminated intravascular thrombosis resulting in multiorgan failure within a short period of time (usually within 1 week).
Other causes. TMA is also well described in transplant recipients, both as a manifestation of transplant rejection and as possible toxicity from immunosuppressive medications such as cyclosporine and tacrolimus. Rarely, this syndrome can be caused by disorders involving regulators of the coagulation cascade (mutations in the gene encoding diacylglycerol kinase epsilon and thrombomodulin)3 and the molecules responsible for vitamin B12 metabolism (mutations in the gene encoding methylmalonic aciduria and homocystinuria type C).4
Advertisement
Evaluation to determine the cause of TMA should initially focus on excluding the aforementioned systemic disorders associated with MAHA and thrombocytopenia. This should be followed by an investigation to distinguish among primary TMA syndromes, including the following:
Kidney biopsy is not helpful for determining cause of primary TMA syndromes and may not distinguish primary TMA syndromes from other disorders such as systemic sclerosis or severe hypertension, which can also manifest the typical pathologic features of TMA.
TTP is a primary TMA syndrome caused by severely reduced activity of the von Willebrand factor-cleaving protease ADAMTS13.5 When ADAMTS13 protease activity is reduced, large von Willebrand factor multimers can accumulate on the endothelial surface of small arterioles and capillaries, leading to potentially fatal platelet accumulation and subsequent mechanical red blood cell fragmentation.5 TTP can be acquired, from an autoantibody inhibitor to ADAMTS13, or it can be hereditary, owing to inherited mutations in ADAMTS13. Genetic testing of the ADAMTS13 gene is the definitive means of documenting the diagnosis of hereditary TTP in an individual with severe ADAMTS13 deficiency (activity < 10% of normal) without an inhibitor. On the other hand, acquired TTP can be confirmed by severe ADAMTS13 deficiency and the presence of an ADAMTS13 autoantibody inhibitor in the appropriate clinical setting (e.g., a patient with MAHA and thrombocytopenia that responds to plasmapheresis).6
Hematologic recovery in TTP can be expected with plasmapheresis, as indicated by improvement in LDH (often within one day) and platelet count (within two to three days).7,8 TTP was ruled out in our patient on the basis of relatively normal ADAMTS13 activity, a negative ADAMTS 13 inhibitor, poor hematologic response to emergency plasmapheresis, and lack of hematologic recovery to suggest resolution of MAHA after three days of plasmapheresis.
Severe hypertension can also cause MAHA and thrombocytopenia with characteristic pathologic features of TMA in the kidneys.9 Grade III/IV retinopathy is more likely to be present, and recovery of kidney function and resolution of MAHA is typical with proper treatment of severe hypertension.9,10 Furthermore, severe hypertension may also occur in primary TMA with severe renal involvement, so the temporal relationship is important.9 Although the patient did have funduscopic findings consistent with grade III hypertensive retinopathy, kidney function and MAHA did not improve despite appropriate management of blood pressure, prompting us to continue our investigation into other causes of TMA.
Shiga toxin-producing pathogens (e.g., Escherichia coli O157:H7) can induce hemolytic uremic syndrome and are often related to exposure to improperly prepared foods, inadequately cooked meats, or farm animals.11 However, our patient did not report any severe abdominal pain or diarrhea with bloody stool, and DNA testing performed on the patient’s stool sample for Shiga-toxin genes was negative. Additional workup investigating non-Shiga-toxin-producing infectious organisms associated with MAHA and thrombocytopenia was also unrevealing, including tests for bacterial (e.g., Clostridioides difficile, Legionella), viral (e.g., cytomegalovirus, hepatitis viruses, human immunodeficiency virus, varicella-zoster virus, Epstein-Barr virus), and fungal (e.g., Aspergillus) infections.
Complement-mediated TMA or atypical hemolytic uremic syndrome is a chronic, systemic, life-threatening disease characterized by TMA and severe multiple end-organ damage caused by dysregulation and uncontrolled activation of the alternative complement pathway. Complement regulatory proteins, including CFH, CFI, and CD46, prevent inappropriate activation of the complement cascade.12 The diagnostic workup for determining cause of complement-mediated TMA includes testing for inhibitory antibodies such as anti-CFH antibody, as well as genetic testing, including evaluation of the CFH, CFI, CD46, CFB, and C3 genes.13 On the other hand, testing for serum complement biomarkers such as CH50, C3, and C4 have no diagnostic role in complement-mediated TMA owing to low sensitivity and specificity. For instance, a low C3 level may only be appreciated in certain mutations such as variants in CFH, CFB, and C3.14
Complement activation is also common in lupus nephritis, and a subset of these patients (17.5%) can present with complement-mediated TMA,15 described as a separate clinical entity that can be resistant to treatment with high-dose corticosteroids, immunotherapy, and plasmapheresis.16 The pathogenesis of renal TMA in lupus nephritis is yet to be determined and may be multifactorial, potentially attributable to antiphospholipid syndrome, malignant hypertension, or scleroderma.17
Our patient had normal plasma levels of complement factors CFH and CFB, along with negative anti-CFH antibody. Complement genetic studies were not obtained, and the patient did not report a family history of TMA disorders suggestive of a hereditary cause. However, one should be mindful that most complement mutations associated with TMA are heterozygous, and many family members with heterozygous mutations are asymptomatic.18 If performed in this patient, complement genetic studies might have provided a more specific diagnosis.
Although complement testing was unrevealing in this patient, a presumptive clinical diagnosis of complement-mediated TMA was made in light of the progressive kidney dysfunction, MAHA, thrombocytopenia, and absence of an alternate explanation for these findings (e.g., no known drug exposure associated with drug-induced TMA, no antiphospholipid syndrome or scleroderma, no response after adequate control of hypertension, no ADAMTS13 deficiency, no Shiga toxin in stool).
The differential diagnosis for the patient’s acute kidney failure included lupus nephritis and TMA.
Plasmapheresis was performed for a total of three days, with no clinical improvement. Then, administration of eculizumab, a complement inhibitor, was commenced in light of the suspected diagnosis of complement-mediated TMA.
C5 inhibitors, including eculizumab and ravulizumab, prevent the cleavage of cell surface-bound complement protein C5 and inhibit generation of proinflammatory terminal complement proteins C5a and the membrane attack complex C5b-9, consequently reducing endothelial cell activation and thrombosis but also increasing the risk of developing meningococcal infections.19 Therefore, in addition to meningococcal vaccination, the patient received antimicrobial prophylaxis with amoxicillin for the duration of eculizumab administration.
Pericardial effusion. During hospitalization, the patient required transfer to the cardiac intensive care unit because of pericardial effusion with tamponade. Pericardiocentesis was performed, and subsequent pericardial fluid studies were suggestive of an exudative effusion.20 There was a predominance of mononuclear cells as well as negative Gram stain, bacterial/fungal cultures, and cytology, favoring active SLE flare as the cause of the exudative pericardial effusion.
Mental status changes. Moreover, the patient’s clinical course was complicated by the development of altered mental status. She became less interactive, could not follow commands, and neurologic examination revealed slightly brisker reflexes in the right upper extremity. This was concerning for possible primary manifestation of neuropsychiatric SLE, as there was past evidence of systemic involvement of SLE.
Other potential etiologies that were considered included infectious encephalitis, given the patient’s immunocompromised state, stroke secondary to central nervous system involvement by SLE, and posterior reversible encephalopathy syndrome, given her recent blood pressure fluctuations with systolic blood pressure higher than 200 mm Hg. Magnetic resonance imaging of the brain was concerning for antibody-mediated striatal encephalitis. This finding is not specific for SLE, and viral encephalitis should also be considered in the differential diagnosis.
A lumbar puncture that was subsequently performed did not show any pleocytosis in the cerebrospinal fluid, rendering an intrathecal infection less likely as the etiology of the patient’s acute encephalopathy and favoring the diagnosis of neuropsychiatric lupus.
Acute kidney injury. The patient’s kidney function continued to deteriorate, requiring kidney replacement therapy within five days of initial presentation to the hospital. Kidney biopsy was considered at this time but was ultimately not performed due to severe thrombocytopenia.
Biopsy of the kidney may be performed for suspected TMA syndrome.21 However, kidney biopsy may not inform treatment decisions for a specific TMA as there is no consensus regarding features on kidney biopsy that definitively diagnose a specific TMA syndrome. Kidney biopsy is not usually done to evaluate patients with acute kidney injury, MAHA, and thrombocytopenia unless there is a specific management decision that would be influenced by the results.
Our patient’s clinical condition was an example of a situation in which a kidney biopsy may have been helpful: it may have identified the extent of ischemic injury sustained in relationship to potential TMA, as well as the degree of irreversible kidney injury to support decision-making for next therapeutic treatment steps. Kidney biopsy may have justified the maintenance or discontinuation of immuno suppressive therapy for lupus nephritis, especially as the patient had not yet developed other systemic manifestations of SLE (e.g., pericardial effusion, neuropsychiatric SLE) at the time kidney biopsy was being considered. The presence of extensive interstitial fibrosis or glomerulosclerosis (class VI advanced sclerosing lupus nephritis), as opposed to focal or diffuse proliferative glomerulonephritis (class III and IV lupus nephritis, respectively), might have prompted discontinuation of immunosuppression.
Although the association between kidney dysfunction and bleeding is well recognized, major complications have been noted to be less than 1%.22 Our patient did not undergo kidney biopsy owing to persistent thrombocytopenia, as thrombocytopenia increases the risk of major complications with kidney biopsy in patients with SLE.23
Intravenous methylprednisolone and mycophenolate mofetil were initially employed as induction therapy for lupus nephritis. Mycophenolate mofetil was preferred over cyclophosphamide because mycophenolate mofetil has been shown to be more effective than intravenous cyclophosphamide in inducing remission of lupus nephritis.24 The patient’s race was a consideration as it has been demonstrated that African American patients with lupus nephritis respond better to mycophenolate mofetil than to intravenous cyclophosphamide (53.9 vs 40%; P = 0.39).25
However, concern for neuropsychiatric SLE with antibody-mediated striatal encephalitis later prompted the discontinuation of mycophenolate mofetil in favor of cyclophosphamide, as cyclophosphamide is the only therapy that has demonstrated efficacy for neuropsychiatric SLE compared with methylprednisolone in a controlled clinical trial.26
Although high-dose glucocorticoids and intravenous cyclophosphamide remain the cornerstone for patients with neuropsychiatric SLE, rituximab or intravenous immunoglobulin may be used if response is not achieved.27 Furthermore, even without studies regarding efficacy of plasmapheresis in patients with lupus antibody-mediated striatal encephalitis, there is some evidence regarding its use in severe neuropsychiatric SLE.28,29 Consequently, plasmapheresis was briefly performed together with administration of cyclophosphamide, with subsequent improvement in the patient’s mental status.
Although the hemolytic anemia and thrombocytopenia resolved after six weeks of therapy with eculizumab, the patient’s kidney function did not recover for more than two months during hospitalization. Eventually the decision was made to stop eculizumab and continue immunosuppressive therapy with cyclophosphamide upon discharge to a rehabilitation facility. Although the patient did not undergo kidney biopsy to guide therapy, the rationale for continued treatment with immunosuppressive therapy at discharge was owing to evidence of other systemic manifestations of SLE during the course of hospitalization, including pericardial effusion and lupus antibody-mediated striatal encephalitis.
The immediate management decision for a patient with suspected TMA is whether to perform plasma exchange for a presumptive diagnosis of TTP or anticomplement therapy for a presumptive diagnosis of complement-mediated TMA.
Plasma exchange should be initiated in adults as soon as diagnosis of TMA is suspected. This helps remove ADAMTS13 autoantibodies, factor H autoantibodies, and hyper-functioning complement components as well as replaces ADAMTS13 and faulty complement regulators.21
Unfortunately, TMA associated with lupus nephritis is usually refractory to treatment with corticosteroids, cyclophosphamide, immunomodulation, and plasma exchange.30,31 However, a 2018 series involving patients with complement-mediated TMA related to SLE reported complete recovery of hematologic parameters in all patients and recovery of kidney function in 80% of patients treated with eculizumab.16
Initial evidence for the efficacy of eculizumab for treating TMA came from a prospective, open-label case-series32 of patients with complement-mediated TMA in which eculizumab improved kidney and hematologic outcomes and reduced thrombotic microangiopathic events including requirement for plasma exchange or infusion, reduction in platelet counts, and initiation of hemodialysis.32
Although C5 inhibitors including eculizumab and ravulizumab are currently the only effective therapy designed to prevent the underlying mechanism of microvascular damage, which is dysregulation of the complement activation system, there are limited data on long-term outcomes in patients with TMA treated with terminal complement inhibition. There are also several unanswered questions regarding C5 inhibitor therapy, including optimal duration of therapy, appropriate strategies for monitoring therapeutic efficacy in patients with suboptimal response or during periods of infection or inflammation, and determining tailored therapeutic regimens in various groups of patients with TMA, including those with complement-mediated TMA.
At Cleveland Clinic, eculizumab therapy has been used broadly, not only for clinically suspected complement-mediated TMA, but also for other TMA syndromes. Owing to the lack of available information regarding management decisions for patients with TMA, investigation into management strategies for these patients is warranted as is research regarding kidney and hematologic outcomes after therapy.
1.Tekin ZE, Yener GO, Yüksel S. Acquired angioedema in juvenile systemic lupus erythematosus: case-based review. Rheumatol Int 2018; 38(8):1577–1584.
2. Kumar N, Surendran D, Bammigatti C. Angioedema as the presenting feature of systemic lupus erythematosus. BMJ Case Rep 2018; 2018:bcr2018224222.
3. Lemaire M, Frémeaux-Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013; 45(5):531–536.
4. Beck BB, van Spronsen F, Diepstra A, Berger RM, Komhoff M. Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol 2017; 32(5):733–741.
5. Zander CB, Cao W, Zheng XL. ADAMTS13 and von Willebrand factor interactions. Curr Opin Hematol 2015; 22(5):452–459.
6. Hassan S, Westwood JP, Ellis D, et al. The utility of ADAMTS13 in differentiating TTP from other acute thrombotic microangiopathies: results from the UK TTP Registry. Br J Haematol 2015; 171(5):830–835.
7. Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 1991; 325(6):393–397.
8. George JN. How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116(20):4060–4069.
9. Van Laecke S, Van Biesen W. Severe hypertension with renal thrombotic microangiopathy: what happened to the usual suspect? Kidney Int 2017; 91(6):1271–1274.
10. Shavit L, Reinus C, Slotki I. Severe renal failure and microangiopathic hemolysis induced by malignant hypertension–case series and review of literature. Clin Nephrol 2010; 73(2):147–152.
11. Allison HE. Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol 2007; 2(2):165–174.
12. Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J 2015; 34(22):2735–2757.
13. Bu F, Maga T, Meyer NC, et al. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol 2014; 25(1):55–64.
14. Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013; 8(4):554–562.
15. Wu LH, Yu F, Tan Y, et al. Inclusion of renal vascular lesions in the 2003 ISN/RPS system for classifying lupus nephritis improves renal outcome predictions. Kidney Int 2013; 83(4):715–723.
16. Park MH, Caselman N, Ulmer S, Weitz IC. Complement-mediated thrombotic microangiopathy associated with lupus nephritis. Blood Adv 2018; 2(16):2090–2094.
17. Song D, Wu L-H, Wang F-M, et al. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther 2013; 15(1):R12.
18. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010; 5(10):1844–1859.
19. Parker CJ, Kar S, Kirkpatrick P. Eculizumab. Nat Rev Drug Discov 2007; 6(7):515–516.
20. Buoro S, Tombetti E, Ceriotti F, et al. What is the normal composition of pericardial fluid? Heart 2021; 107(19):1584–1590.
21. Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney.Clin J Am Soc Nephrol 2018; 13(2):300–317.
22. Tøndel C, Vikse BE, Bostad L, Svarstad E. Safety and complications of percutaneous kidney biopsies in 715 children and 8573 adults in Norway 1988–2010. Clin J Am Soc Nephrol 2012; 7(10):1591–1597.
23. Chen TK, Estrella MM, Fine DM. Predictors of kidney biopsy complication among patients with systemic lupus erythematosus. Lupus 2012; 21(8):848–854.
24. Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 2005; 353(21):2219–2228.
25. Isenberg D, Appel GB, Contreras G, et al. Influence of race/ethnicity on response to lupus nephritis treatment: the ALMS study. Rheumatology (Oxford) 2010; 49(1):128–140.
26. Barile-Fabris L, Ariza-Andraca R, Olguín-Ortega L, et al. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann Rheum Dis 2005; 64(4):620–625.
27. Magro-Checa C, Zirkzee EJ, Huizinga TW, Steup-Beekman GM. Management of neuropsychiatric systemic lupus erythematosus: current approaches and future perspectives. Drugs 2016; 76(4):459–483.
28. Kovacs B, Lafferty TL, Brent LH, DeHoratius RJ. Transverse myelopathy in systemic lupus erythematosus: an analysis of 14 cases and review of the litrature. Ann Rheum Dis 2000; 59(2):120–124.
29. Bartolucci P, Brechignac S, Cohen P, Le Guern V, Guillevin L. Adjunctive plasma exchanges to treat neuropsychiatric lupus: a retrospective study on 10 patients. Lupus 2007; 16(10):817–822.
29. Wu LH, Yu F, Tan Y, et al. Inclusion of renal vascular lesions in the 2003 ISN/RPS system for classifying lupus nephritis improves renal outcome predictions. Kidney Int 2013; 83(4):715–723.
30. Yu F, Haas M, Glassock R, Zhao M-H. Redefining lupus nephritis: clinical implications of pathophysiologic subtypes. Nat Rev Nephrol 2017; 13(8):483–495.
31. Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013; 368(23):2169–2181.
Advertisement

Pediatric urologists lead quality improvement initiative, author systemwide guideline

Fixed-dose single-pill combinations and future therapies

Reproductive urologists publish a contemporary review to guide practice

Two recent cases show favorable pain and cosmesis outcomes

Meta-analysis assesses outcomes in adolescent age vs. mid-adulthood

Proteinuria reduction remains the most important treatment target.

IgA nephropathy is a relatively common autoimmune glomerular disease that can be diagnosed only by biopsy

Oncologic and functional outcomes are promising, but selection is key