Locations:

Pathophysiology of hyperphosphatemia
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/1353c13d-eb04-4222-a25b-6f49b04b7d02/650x450-Phosphorus-Kideny-Disease_jpg)
650×450-Phosphorus-Kideny-Disease
By Georges N. Nakhoul, MD, Stacey Jolly, MD, MAS, FACP, and Hernan Rincon-Choles, MD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
The balance between dietary intake and excretion of phosphorus can be impaired in patients with decreased renal function, leading to hyperphosphatemia. Many patients with end-stage renal disease on dialysis require phosphorus-binding drugs to control their serum phosphorus levels.
In this review, we discuss the pathophysiology of hyperphosphatemia in kidney disease, its consequences, and how to control it, focusing on the different classes of phosphorus binders.
With kidney disease common and on the increase, nephrologists and internists need to work together to provide optimal care.
Further, many internists in managed care plans and accountable care organizations now handle many tasks previously left to specialists — including prescribing and managing phosphorus binders in patients with kidney disease.
The pathophysiology of bone mineral disorders in kidney disease is complex. To simplify the discussion, we will address it in three parts:
Phosphorus is a macronutrient essential for a range of cellular functions that include structure, energy production, metabolism and cell signaling. It exists primarily in the form of inorganic phosphate.
Net phosphorus balance depends on dietary phosphorus intake, gastrointestinal absorption, renal function and flux between extracellular and intracellular (skeletal) pools.
An average Western diet provides 20 mg of phosphorus per kilogram of body weight per day. Of this, 13 mg/kg is absorbed, and the rest is excreted in the feces.
Advertisement
Absorption of dietary phosphorus occurs mainly in the jejunum. It is mediated by both a paracellular sodium-independent pathway (driven by high intraluminal phosphorus content) and by active sodium-dependent cotransporters. It is also influenced by diet and promoted by active vitamin D (1,25 dihydroxyvitamin D3, also called calcitriol).
Absorbed phosphorus enters the extracellular fluid and shifts in and out of the skeleton under the influence of parathyroid hormone.
Phosphorus excretion is handled almost entirely by the kidneys. Phosphorus is freely filtered at the glomerulus and reabsorbed mainly in the proximal tubule by sodium-phosphate cotransporters.
Normally, when phosphorus intake is adequate, most of the filtered phosphorus is reabsorbed and only 10 to 20 percent is excreted in the urine. However, the threshold for phosphorus reabsorption in the proximal tubule is influenced by parathyroid hormone, FGF23, and dietary phosphorus intake: low serum phosphate levels lead to an increase in the synthesis of sodium-phosphorus cotransporters, resulting in increased (nearly complete) proximal reabsorption and an increase in the serum phosphorus concentration. Conversely, both parathyroid hormone and FGF23 are phosphaturic and decrease the number of phosphorus transporters, which in turn leads to increased phosphorus excretion and a decrease in serum phosphorus concentration.
FGF23 is a phosphaturic glycoprotein secreted by osteoblasts and osteocytes. It acts by binding to fibroblastic growth receptor 1 in the presence of its coreceptor, the Klotho protein.
Advertisement
FGF23 is regulated by serum phosphorus levels and plays a major role in the response to elevated serum phosphorus. It causes a direct increase in urinary phosphorus excretion, a decrease in intestinal phosphorus absorption (indirectly via inhibition of calcitriol), and decreased bone resorption via a decrease in parathyroid hormone production.
Parathyroid hormone, in contrast, has a mixed effect. It increases renal excretion of phosphorus on one hand but increases phosphorus release from bone into the serum on the other. The latter is accomplished by increasing both bone resorption (directly) and intestinal absorption (indirectly, via stimulation of calcitriol) of phosphorus.8
FGF23 inhibits parathyroid hormone and calcitriol. Parathyroid hormone stimulates both FGF23 and calcitriol, whereas calcitriol inhibits parathyroid hormone.The complex interplay between these hormones is shown below.
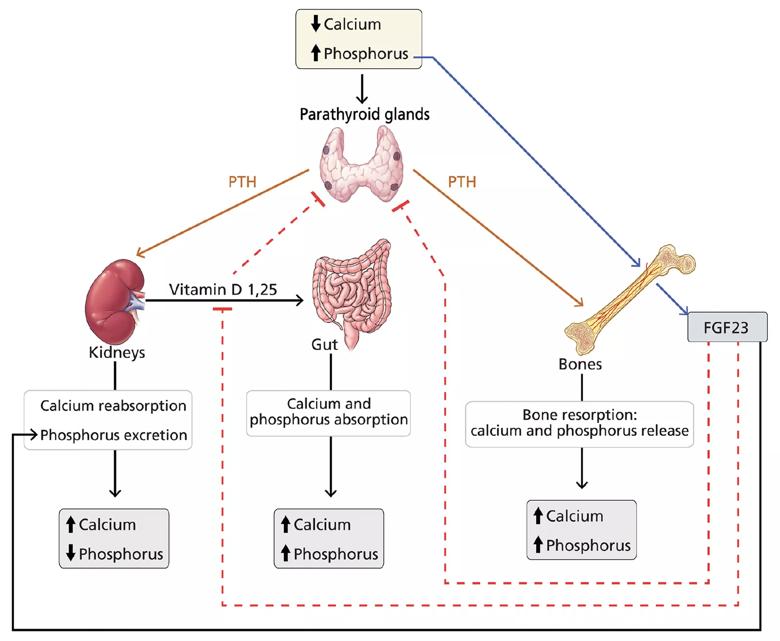
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/bd5b1152-ad97-455f-a052-62e826d93ff5/sekar_phosphorusbinders_f1_jpg)
Figure 1. Hormonal regulation of calcium and phosphorus. Serum calcium and phosphorus balance is maintained by a tight interplay between parathyroid hormone (PTH), vitamin D and fibroblast growth factor 23 (FGF23).
In chronic kidney disease, phosphorus retention can trigger secondary hyperparathyroidism, as rising phosphorus levels stimulate FGF23. In the early stages of chronic kidney disease, this response can correct the phosphorus levels, but with several consequences:
Advertisement
As chronic kidney disease progresses, the glomerular filtration rate falls, the phosphorus level rises, and the above sequence of events is repeated and accentuated, which leads to correction of the phosphorus levels. However, once the glomerular filtration rate falls below 25 to 40 mL/min/1.73 m2, these response mechanisms no longer suffice and the phosphorus level stays elevated.
As the elevated phosphorus level is likely to be the triggering event behind secondary renal hyperparathyroidism, it needs to be controlled. This is accomplished by restricting dietary phosphorus and using phosphorus binders.
In caring for patients with chronic kidney disease, it is important to prevent and treat hyperphosphatemia with a combination of dietary restrictions and phosphorus binders. This post reviews the pathophysiology of hyperphosphatemia. Future posts will discuss its control and the different classes of phosphorus binders with respect to their availability, cost, side effects and scenarios in which one class of binder may be more beneficial than another.
This abridged article was originally published in Cleveland Clinic Journal of Medicine.
Dr. Rincon-Choles is staff in the Department of Nephrology and Hypertension. Dr. Jolly is staff in the Department of Internal Medicine. Dr. Nakhoul is Director of the Center for Chronic Kidney Disease in the Department of Nephrology and Hypertension.
Advertisement
Advertisement

Findings underscore the importance of a multifaceted prevention strategy

Innovative single-port technique results in no visible scarring

The new application that could augment cancer detection, improve efficiency

Pediatric urologists lead quality improvement initiative, author systemwide guideline

Fixed-dose single-pill combinations and future therapies

Reproductive urologists publish a contemporary review to guide practice

Two recent cases show favorable pain and cosmesis outcomes

Meta-analysis assesses outcomes in adolescent age vs. mid-adulthood