Locations:

Defect in myosin binding may explain why a significant percentage of patients do not adequately respond to anti-TNF therapy

By Elaine Husni, MD, MPH and Unni Chandrasekharan, PhD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Although tumor necrosis factor (TNF) inhibiting agents have revolutionized our treatment of patients with rheumatoid and psoriatic diseases, up to 40% of patients do not respond or only partially respond, or lose efficacy of therapeutic response over time. We need to better understand how to provide personalized care for patients to improve quality of life and clinical outcomes rather than the current trial-and-error approach.
There are currently no clinical tests available to predict responsiveness to anti-TNF therapy in advance. Small studies have supported that patients carrying a TNFR2-M196R polymorphic variant demonstrate a higher risk of being an inadequate responder to anti-TNF drugs; however, the underlying mechanisms are unclear. We have shown previously that non-muscle myosin (myosin) functions as a negative regulator of TNFR2 activation. In a recent study, presented at the annual meeting of the American College of Rheumatology in November 2019, we tested the hypothesis that a defect in myosin binding to TNFR2-M196R causes TNF-independent proinflammatory activity, which leads to reduced responsiveness to anti-TNF agents. The ability to genotype this polymorphism will enable us to predict inadequate responders to anti-TNF therapies early on, and help develop a personalized approach to the treatment of these immune-mediated diseases.
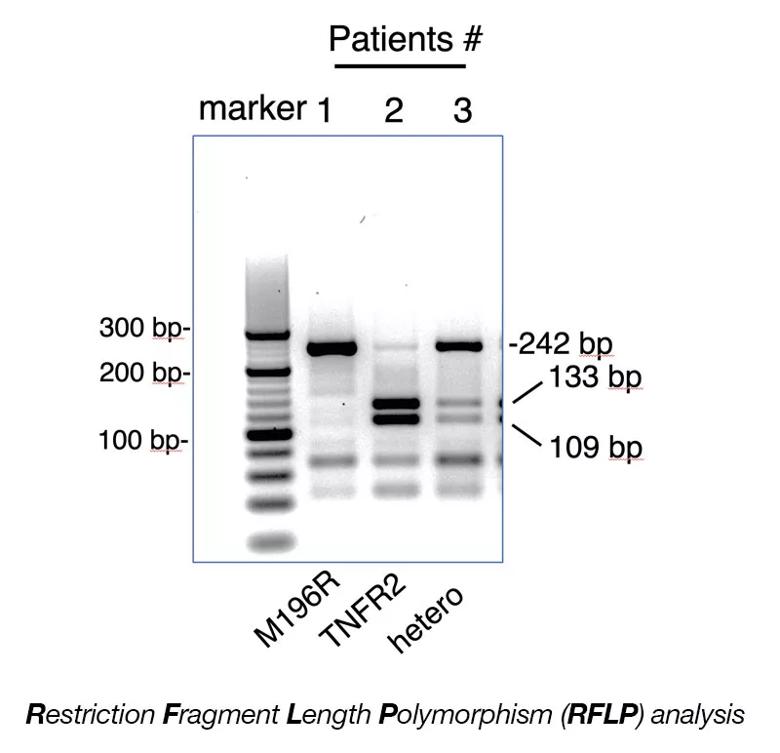
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/351df4ad-5454-41f5-956f-eb3225743feb/20-RHE-1877572-Husni_Myosin-defect-Inset_800x772_jpg)
RFLP analysis was performed on DNA isolated from the buffy coat of human subjects using the PCR. NlaIII restriction enzyme is used to digest the 242 base pair (bp) PCR product (see reference below). Lane 1: uncut 242 bp fragment corresponds to homozygous TNFR2-M196R. Lane 2: NlaIII cut fragments (133bp and 109bp) correspond to homozygous for TNFR2. Lane 3: Uncut 242 bp and cut 133bp and 109 bp correspond to TNFR2/TNFR2-M196R hemizygous. Ref: Al-Ansari et. al., Tissue Antigens 55, 97-99 (2000).
Advertisement
Through co-immunoprecipitation studies, we demonstrated myosin binds to endogenous TNFR2, but not TNFR2-M196R in leukocytes isolated from patients’ blood. We also found TNFR2-M196R expressing cells show TNF-independent, Rho Kinase 1 (ROCK1) activity and proinflammatory gene induction (e.g., ICAM1, CXCL10) in cultured cells. Importantly, a TNF-neutralizing antibody failed to inhibit proinflammatory gene induction in TNFR2-M196R expressing cells. Furthermore, this elevated proinflammatory gene induction upon TNF is sustained for a longer time period in TNFR2-M196R expressing cells is (8 -10 hours) compared with TNFR2 expressing cells (4-6 hours).
Mechanistically, TNFR2-M196R drives cells to a constitutive proinflammatory state, potentially due to a defect in myosin binding. Since this constitutive activity is TNF-independent, and given that approximately 20% of the general population carry the TNFR2-M196R polymorphic variant, our findings may potentially explain, in part, why a significant percentage of patients do not adequately respond to anti-TNF therapy.
Our mechanism-based approach implicating TNFR2-M196R testing in patients that are candidates for anti-TNF therapy may not only help to predict anti-TNF responsiveness but may also reveal novel pathways that can be targeted to treat inadequate responders of anti-TNF therapy in a subset of patients with rheumatoid and psoriatic diseases. Given the higher expense, longer time, potential serious adverse effects and emotional toll on patients in cycling through multiple disease modifying agents, identifying genetic biomarkers of treatment response would change our existing practice and facilitate a more personalized treatment approach.
Advertisement
Advertisement

First full characterization of kidney microbiome unlocks potential to prevent kidney stones

Researchers identify potential path to retaining chemo sensitivity
Large-scale joint study links elevated TMAO blood levels and chronic kidney disease risk over time
Investigators are developing a deep learning model to predict health outcomes in ICUs.

Preclinical work promises large-scale data with minimal bias to inform development of clinical tests

Cleveland Clinic researchers pursue answers on basic science and clinical fronts

Study suggests sex-specific pathways show potential for sex-specific therapeutic approaches

Cleveland Clinic launches Quantum Innovation Catalyzer Program to help start-up companies access advanced research technology