Locations:

Case illustrates mortality of severe disease
By Soumya Chatterjee, MD, MS, FRCP
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
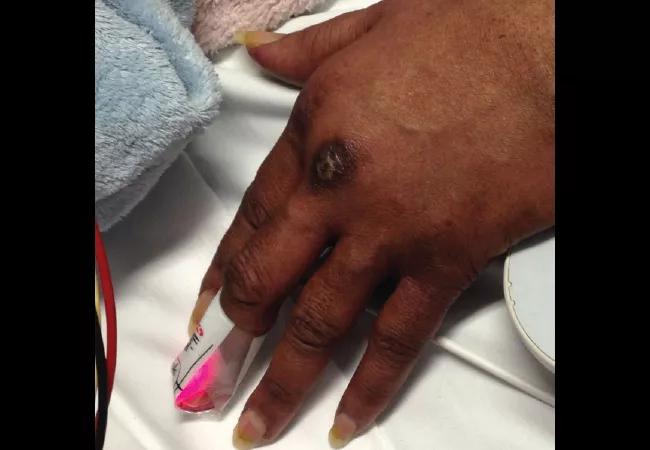
A 58-year-old female was admitted with shortness of breath and a rash that had been present for three weeks. On examination, she had swollen eyelids with a violaceous tinge; erythema and focal erosions of neck and submental chin, as well as upper chest; and violaceous plaques overlying the metacarpophalangeal (MCP) joints of her hands (Gottron papules). She was also severely short of breath requiring supplemental oxygen. She denied fever, but had a cough with minimal expectoration of clear phlegm. There was no clinical synovitis, but she was mildly weak in her proximal muscles. She had diffuse crackles at the lung bases, up to the mid-zones.
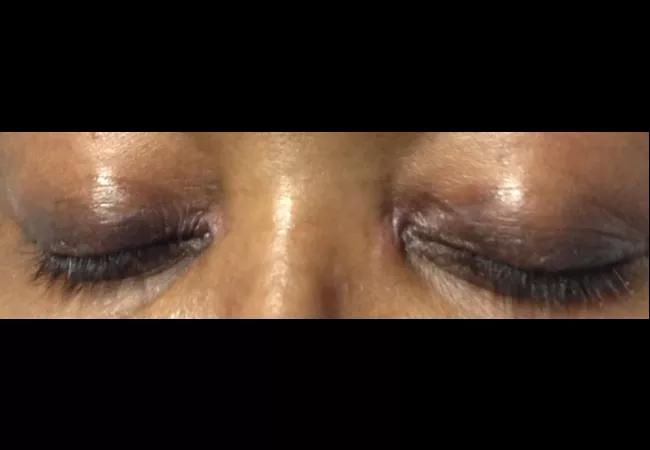
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/8e9a7f0a-7fd1-4f49-af00-af56b1f8ca57/17-RHE-1225-Chatterjee-Inset-Image-03-650pxl-width_jpg)
Laboratory investigations included a negative antinuclear antibody (ANA), antibodies to extractable nuclear antigens (except a low titer anti-U1-RNP antibody), and the antisynthetase antibody panel. Her creatine kinase was 350 U/L (Reference range: 30-220 U/L) and serum ferritin was 1237 ng/mL (Reference range: 18-300 ng/mL). Pulmonary function tests showed severe restrictive lung disease with forced vital capacity 63 percent of predicted and transfer factor (DLco) 33 percent of predicted. Thoracic high resolution computerized tomography (HRCT) showed evidence of severe interstitial lung disease (ILD) (organizing pneumonia and/or interstitial pneumonitis) mostly involving mid to lower lung zones. These findings were suggestive of dermatomyositis (DM) with severe ILD.
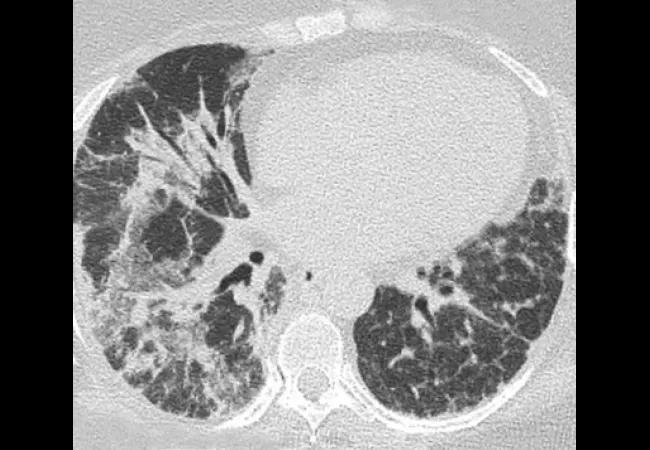
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/c226bd1d-03e9-4acb-8d1d-9741a2f315bb/17-RHE-1225-Chatterjee-Inset-Image-02-650pxl-width_jpg)
In the next few weeks some of the Gottron papules became ulcerated.
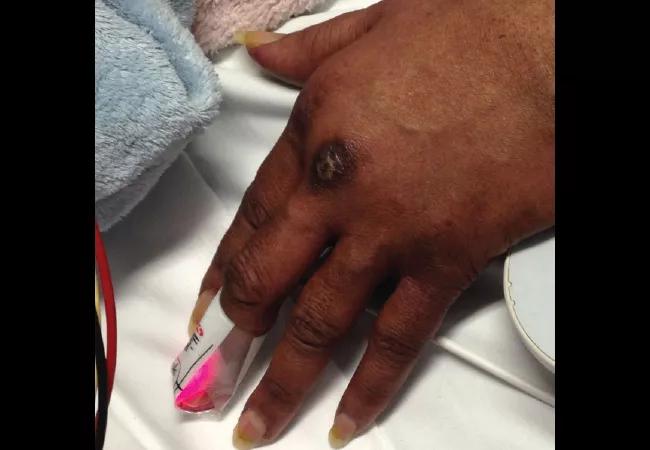
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/704bd2e4-9830-4fbb-a605-75dfc7087323/17-RHE-1225-Chatterjee-Inset-Image-01-650pxl-width_jpg)
She also developed focal and erythematous tender papules of the palms and some fingers. Based on these findings, she matched the clinical phenotype of melanoma differentiation associated protein 5 (MDA-5) dermatomyositis, the diagnosis of which was confirmed when the MDA-5 antibody came back positive.
Advertisement
Once infections were ruled out, she was treated with high-dose intravenous methylprednisolone followed by oral prednisone 60 mg daily. Unfortunately, cyclophosphamide and mycophenolate were denied by her insurance. Based on some published data on MDA-5 DM, she was started on oral tacrolimus. However, she did not notice any improvement in her breathing, though her skin disease stabilized. With worsening hypoxia, her home oxygen was titrated up from 3 L/min to 6 L/min. In spite of a good appetite, she kept losing weight.
While her breathing continued to deteriorate, serial thoracic HRCT scans revealed areas of ground glass opacification being replaced by interstitial thickening and dense fibrosis, and eventual progression to diffuse reticular opacities associated with architectural distortion and traction bronchiolectasis. She underwent bronchoscopy with broncho-alveolar lavage (BAL) and transbronchial lung biopsy. Lung biopsy showed reactive type 2 cells with scattered areas of intra-alveolar fibrin and areas of organizing inflammation. Neutrophils were present both within the septal capillaries as well as in some areas within the alveolar space.
Ultimately, hypoxia requiring positive pressure ventilation necessitated admission to the intensive care unit. Though BAL was negative for opportunistic infections, broad spectrum antibiotics were started. Oxygen requirements kept rising. Finally, she showed evidence of multi-organ failure with thrombocytopenia, transaminitis and hematuria. Despite escalation of oxygen therapy, her respiratory status continued to decline, and she developed hypotension requiring vasopressors. She ultimately died of cardiac arrest secondary to respiratory failure.
Advertisement
In 2005, Sato et al identified a novel autoantibody recognizing a 140 kDa protein in patients with clinically amyopathic DM (CADM, initially termed CADM-140). The 140 kDa autoantigen was subsequently identified as melanoma differentiation associated protein 5 (MDA-5). In the initial studies in Japanese cohorts, most patients were clinically amyopathic, and often had rapidly progressive ILD. Ultimately, Fiorentino et al linked the unusual cutaneous findings of CADM (ulceration and palmar papules) with ILD and described its association with anti-MDA-5 antibody.
More recently, based on observational cohort studies, further detailed descriptions of MDA-5 dermatomyositis have become available. The clinical phenotype represents an overlap of a severe form of vasculopathy and a rapidly progressive ILD. Unique findings that differentiate MDA-5 DM from classical DM include little or no myositis, skin ulceration (affecting lateral nail folds, Gottron papules and elbows), tender palmar papules and severe ILD. Other features include weight loss, oral pain and/or ulceration, mechanic’s hands, hand edema, polyarthritis/arthralgia and diffuse alopecia. Hyperferritinemia and negative ANA are common. When lung disease is severe, there may be variable response to mycophenolate and tacrolimus along with high dose glucocorticoids. Other therapies with some reported success include basiliximab and plasmapheresis. It has been found that if MDA-5 DM patients have a favorable response to an initial treatment, they may have a better long-term outcome.
Advertisement
There have been significant differences in the phenotype of lung disease between the published Japanese cohorts and those reported in the U.S. Japanese patients quite often have an unfavorable pulmonary outcome. However, in the US, the prognosis has been more variable. Some patients with severe immunosuppressant-refractory ILD (including our case) have died from rapidly progressive respiratory failure while waiting for a lung transplant. Yet other patients have a relatively milder course and respond favorably to immunosuppressant agents. It is not clear why this difference is seen in these different patient populations. It has been proposed that there may be differences in host genetic risk factors and environmental factors that are novel and yet undefined.
Dr. Chatterjee directs the Scleroderma Program in the Department of Rheumatic and Immunologic Diseases.
Advertisement
Advertisement

The case for continued vigilance, counseling and antivirals

High fevers, diffuse rashes pointed to an unexpected diagnosis

No-cost learning and CME credit are part of this webcast series

Summit broadens understanding of new therapies and disease management

Program empowers users with PsA to take charge of their mental well being

Nitric oxide plays a key role in vascular physiology

CAR T-cell therapy may offer reason for optimism that those with SLE can experience improvement in quality of life.

Unraveling the TNFA receptor 2/dendritic cell axis