Locations:

An inability to obtain a biopsy of the patient’s hilar lymphadenopathy due to severe PAH and severe hypoxemia made diagnosis verification challenging
Written by Eduardo Mireles-Cabodevila, MD, and Jaime Vondenberg, DO
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
A 65-year-old-man was referred to Cleveland Clinic’s Medical Intensive Care Unit (MICU) for emergent evaluation for lung transplantation. The patient was diagnosed with idiopathic pulmonary hypertension three years prior to admission. Therapy for this had escalated to an infusion of treprostinil, oral tadalafil and macitentan. He developed worsening hypoxemia and features of right heart failure that led to admission to another facility. The patient was referred to Cleveland Clinic for accelerated transplant evaluation as the course of the disease progressed. On arrival to the MICU, the patient required a 100% high-flow nasal cannula to maintain oxygen saturations above 90%. With effort or cough, the patient would desaturate, and he had to be placed on an additional oxygen face mask.
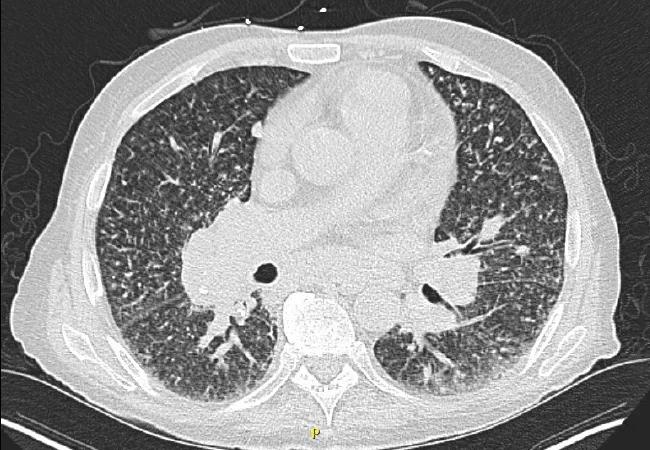
There were several atypical features related to idiopathic pulmonary hypertension. They had periungual erythema and periorbital and forearm violaceous, erythematous and edematous dermatitis. A CT of the chest (Figure 1) revealed hilar/mediastinal lymphadenopathy, multiple bilateral pulmonary nodules, chronic interstitial prominence and interlobular thickening suggestive of pulmonary veno-occlusive disease (PVOD)/pulmonary capillary hemangiomatosis. The initial laboratories demonstrated hypereosinophilia (2700 eosinophils /uL). The team involved consultants from the advanced lung disease service, dermatology, rheumatology and immunology.
A pulmonary artery catheter revealed pulmonary hypertension (mean 52 mmHg) with normal right atrial pressure (6 mmHg), normal pulmonary capillary occlusion pressure (16 mmHg) but an elevated cardiac index (3.1 L/min/m2). This was suggestive of vasodilator therapy overdose, so treprostinil was weaned down. The patient’s hypoxemia and hypereosinophila worsened and peaked at 3800 eosinophils/uL (Figure 1). Steroids were prescribed while further workup was done.
Advertisement
Testing did not reveal any allergic/infectious etiology, and skin biopsy was not consistent with connective tissue disease. Rheumatologic serologies were notable for cANCA positivity, but PR-3/MPO and ANA by IFA were both negative. RNP antibody was elevated at 4.8 AI, IgE was elevated at 2,243 kU/L, IgG4 was elevated at 2,434 mg/dL, and low complement C3 level 53mg/dL and low complement C4 level < 2mg/dL. These serologies, in addition to the patient’s clinical presentation, were not consistent with eosinophilic granulomatosis with polyangiitis, idiopathic inflammatory myopathies, antisynthetase syndrome or scleroderma sine sclerosis. Given that the antibody profile did not appear consistent with a unifying diagnosis, there was a concern for infection or malignancy so a PET-CT from skull to thigh was ordered. The PET-CT was notable for FDG-avid uptake of the thyroid gland, most notably the right side of the thyroid, but without discrete nodules.
In the context of the patient’s severe pulmonary arterial hypertension, CT findings with hilar lymphadenopathy as well as reticulonodular and ground-glass opacities in both lungs, the concern for PVOD, elevated IgG4, hypereosinophilia and low C3/C4 — all of which had been responsive to steroid therapy — IgG4 related disease (IgG4-RD) appeared to be the most likely unifying diagnosis. (Figure 2)
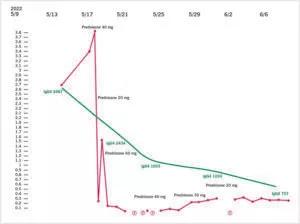
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/54927fcc-0325-44c1-bbc7-39ecd9717b50/22-PUL-3153122-CQD-Complex-Case-2-Dr_-Eduardo-Mireles805X601-300x224_jpg)
Figure 2. Eosinophil and IgG4 levels and its relation with steroid dosing
IgG4-RD is characterized by fibroinflammatory infiltrate and the ability to affect any organ system, though with a predilection for the endocrine system. Pathology of IgG4-RD consists of lymphoplasmacytic infiltrate, which, on further immunohistochemistry staining, reveals IgG4 deposition. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD include three criteria:
Advertisement
Other findings that can be seen in IgG4-RD include hypereosinophilia, hypocomplementemia and an elevated IgE level — all of which were seen in this case. IgG4-RD is generally very steroid responsive, as was also demonstrated in this case.
Ideally in the diagnosis of IgG4-RD, tissue diagnosis would also be completed to verify the diagnosis. The main limiting factor, in this case, was the inability to obtain a biopsy of the patient’s hilar lymphadenopathy due to severe PAH and severe hypoxemia. For this reason, we discussed with the patient obtaining a biopsy of the thyroid gland to further support the suspected diagnosis of IgG4-RD. Cytopathology of the thyroid revealed chronically inflamed fibrous tissue and Hurthle cells both of which can be seen in the sclerosing variant of Hashimoto’s thyroiditis and IgG4-RD. Unfortunately, in this case, IgG4 staining was limited due to high background staining/fine needle aspiration as opposed to open excision and could not provide a definitive diagnosis of IgG4-RD. Despite the limitations of the fine needle aspiration, the cytology findings in addition to elevated TPO and thyroglobulin antibodies were supportive of the suspected IgG4RD diagnosis. The patient was discharged after 27 days, only with oxygen with exertion. Therapy with prednisone 20mg daily was continued for further follow-up with our pulmonary and rheumatology departments.
Advertisement
Advertisement

Takeaways from the most recent annual meeting centered around clinical advances, AI integration and professional development

Recent breakthroughs have brought attention to a previously overlooked condition

A review of treatment options for patients who may not qualify for surgery

Looking at the real-world impact and the future pipeline of targeted therapies

The progressive training program aims to help clinicians improve patient care

New breakthroughs are shaping the future of COPD management and offering hope for challenging cases

Exploring the impact of chronic cough from daily life to innovative medical solutions

How Cleveland Clinic transformed a single ultrasound machine into a cutting-edge, hospital-wide POCUS program