Locations:

Case highlights unusual presentation
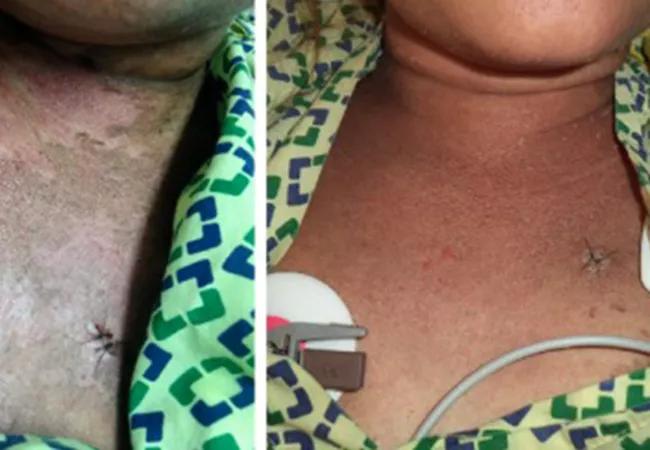
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/e5abd41a-f616-42db-82ed-b873b211dcea/650x450_jpg)
650×450
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
Adult Onset Still’s Disease (AOSD) is a rare systemic inflammatory disorder characterized by the following triad of signs and symptoms: persistent fevers, joint pain and a transient, salmon-colored rash. The cause of the disease is unknown, but it is thought to be an autoinflammatory disorder triggered by infectious agents. AOSD may share similar very high levels of ferritin as encountered in hemophagocytic lymphohistiocytosis (HLH) — a rare but possibly fatal disease presenting with overactive histiocytes and lymphocytes.
This unique case report, recently published in Case Reports in Medicine, highlights a patient who presents with symptoms of these two rare diseases.
Two months after moving from Asia to the United States, a 28-year-old woman developed pain and stiffness in the metacarpophalangeal and proximal interphalangeal joints of both hands. Her medical history was unremarkable, and she was taking no prescription or over-the-counter drugs. An erythematous skin rash quickly followed her joint pain, spreading over her entire body.
She was diagnosed with an allergy at an outpatient clinic and received oral antihistamines, but with no improvement. In fact, the patient continued to worsen and developed additional symptoms of dyspnea, chills, fever and cough. She was admitted to an outside hospital for respiratory failure. After 14 days, she was transferred to Cleveland Clinic.
Upon arrival, the patient was febrile, hypotensive and had a diffuse scaly rash, periorbital edema and icterus. Laboratory data revealed anemia, leukocytosis, thrombocytopenia, elevated liver enzymes, hypertriglyceridemia, increased interleukin-2 receptor levels and very high ferritin levels.
Advertisement
Bone marrow biopsy showed hemophagocytosis, and skin biopsy was suggestive of Still’s disease.
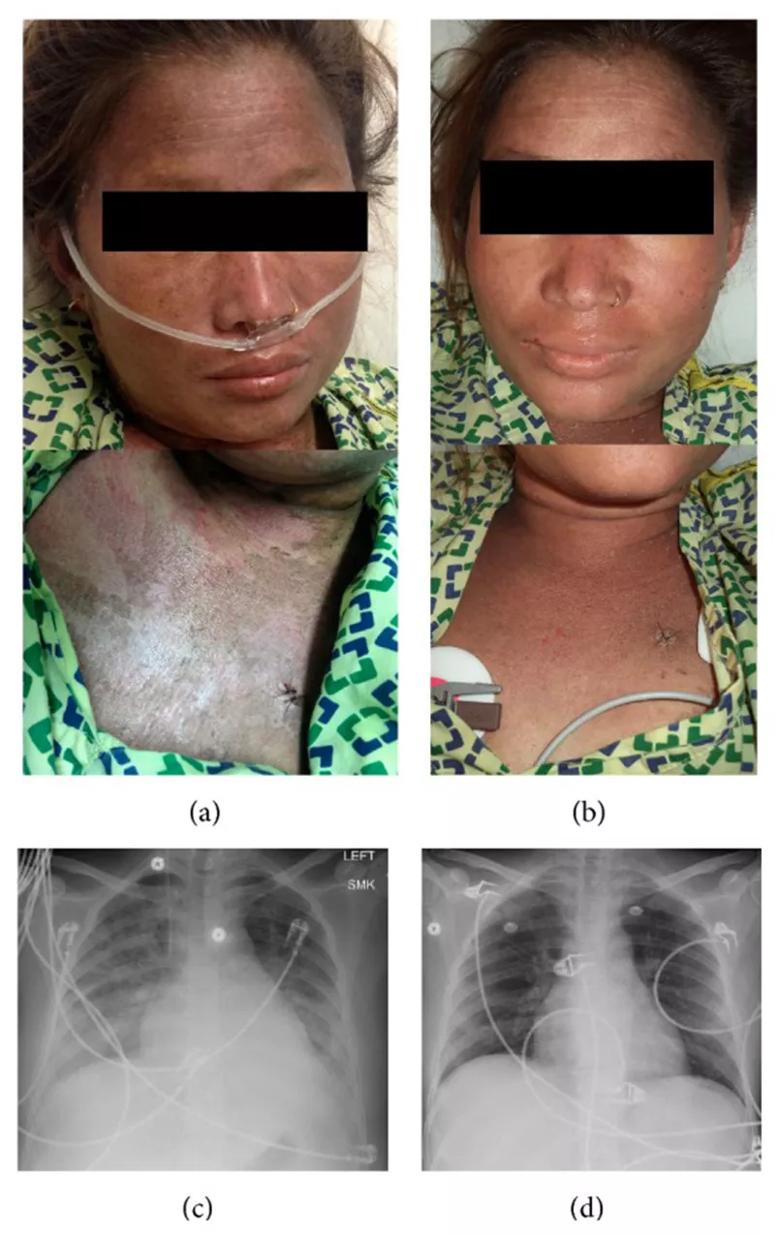
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/a271e96f-59dc-436c-8848-e996db972267/805x-Inset_jpg)
Photograph of the rash and chest X-ray at presentation and discharge. (a) Face and neck upon presentation. (b) Face and neck upon discharge. (c) Chest X-ray at presentation showing bilateral infiltrates. (d) Chest X-ray at discharge showing resolution of infiltrates.
In summary: Both. The patient presented with all four major and three of the four minor criteria for AOSD. She also presented with six out of eight criteria for HLH.
Upon diagnosis, the patient immediately began treatment with daily doses of methylprednisolone. The fever lessened and the rash improved. Due to these improvements, the dose of methylprednisolone was reduced. However, shortly after reducing the dose of steroids, the patient developed a high fever of 40.5°C and increased shortness of breath. The patient was transferred to the ICU, intubated and given anakinra (an IL-2 receptor antagonist) along with prednisone. The patient’s symptoms drastically improved. She was discharged six days later on cyclosporine and a gradual prednisone taper.
This patient case highlights the overlapping symptomatology and pathogenesis of AOSD and HLH that can make diagnosis complicated. Given the potentially fatal course of HLH, early identification and treatment are crucial to providing patients with either disease a positive outcome. This case also reminds us that rare conditions should be kept in the differential diagnosis of unusual presentations.
Advertisement
Dr. Brateanu is staff in the Department of Internal Medicine.
Advertisement
Advertisement

Emerging evidence suggests a patient-specific approach

Not if they meet at least one criterion for presumptive evidence of immunity

Essential prescribing tips for patients with sulfonamide allergies

Confounding symptoms and a complex medical history prove diagnostically challenging

An updated review of risk factors, management and treatment considerations
OMT may be right for some with Graves’ eye disease

Perserverance may depend on several specifics, including medication type, insurance coverage and medium-term weight loss

Abstinence from combustibles, dependence on vaping