Locations:

Case study underscores need to factor in a rare syndrome
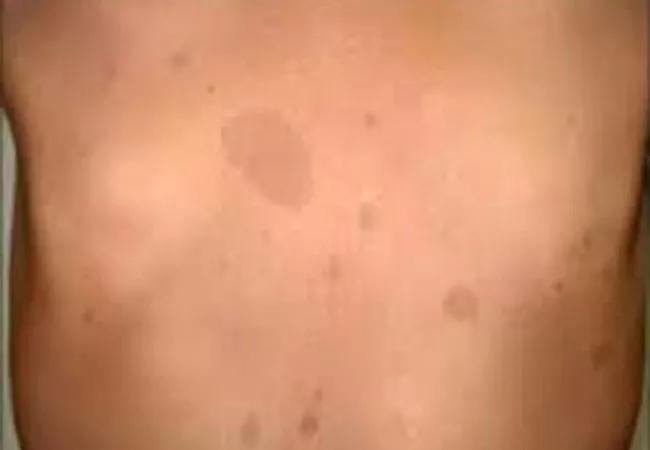
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/be334c33-aad3-4bbf-bb3b-4c65b39bbcd9/19-NEU-430-cafe-au-lait-650x450_jpg)
19-NEU-430-cafe-au-lait-650×450
By Manikum Moodley, MD, FCP, FRCP
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
A 19-month-old boy was referred to Cleveland Clinic’s pediatric neurology clinic due to concern that he had neurofibromatosis type 1 (NF1). His past medical history was significant for macrocephaly and plagiocephaly. Birth and development had been normal. His family history was positive for epilepsy, learning difficulties and café-au-lait macules (CALMs) in his mother, but she had no additional features of NF1. His father had a history of bipolar disorder, learning difficulties and CALMs.
Physical examination revealed café-au-lait modules (CALMs), mainly on the trunk. There were more than six CALMs (the threshold number in diagnostic criteria for NF1), each more than 5 mm in diameter at the widest point. The boy’s parents said he’d had CALMs since 14 months of age. Neurological examination was normal, and there was no evidence of neurofibromas, Lisch nodules or other tumor manifestations.
The patient’s reported family history was negative for known or suspected genetic disease, birth defects, malformation syndromes, chromosomal disorders, metabolic disorders, developmental delay, recurrent pregnancy loss, stillbirth, unexplained infant death and consanguinity.
Cardiac evaluation, including ECG and echocardiogram, revealed no abnormalities. Orthopaedic evaluation was positive for benign external rotation deformity of both hips.
CALMs are common in children (sample photo above), and they most often present as one or two brown spots in an otherwise healthy child. However, the presence of multiple CALMS without other cutaneous anomalies should raise the possibility of an associated syndrome. Of these syndromes, NF1 is the most common. However, Legius syndrome also presents with multiple CALMs.
Advertisement
Initially described as an NF1-like syndrome, Legius syndrome is an autosomal dominant disorder that shares clinical features with NF1, including multiple CALMs with or without axillary freckling and macrocephaly.1 However, it lacks the severe features of NF1, such as Lisch nodules of the iris, neurofibromas, optic pathway gliomas and increased risk for malignancies.2 Definitive diagnosis is necessary since it is associated with a better prognosis relative to NF1, but clinical data for Legius syndrome remain limited.
Because of the absence of neurofibromas, Lisch nodules and other tumor manifestations, our patient’s clinical findings were insufficient to make a diagnosis of NF1, but such a diagnosis could not be ruled out completely.
The parents desired to either confirm or rule out an NF1 diagnosis, so an MRI of the brain was taken at 21 months of age. It showed no definitive findings consistent with NF1.
Legius syndrome was suspected, and genetic testing was recommended. After extensive counseling on the benefits and limitations of such testing, the parents agreed to proceed. Genetic testing of the patient showed a heterozygous truncating mutation in exon 7 of the SPRED1 gene, which confirmed the diagnosis of Legius syndrome.
On a subsequent visit, the patient’s mother underwent genetic testing for Legius syndrome and was found to have the same mutation in the SPRED1 gene, confirming a diagnosis of Legius syndrome for her as well. She was told each child she has will have a 50% chance of having Legius syndrome and that genetic counseling would be appropriate before or during any future pregnancies.
Advertisement
The first case of Legius syndrome was reported by Eric Legius and colleagues in 2007.1 The syndrome is caused by a heterozygous germline loss-of-function mutation in the SPRED1 gene,1 which encodes a negative regulator of the Ras-MAP kinase signaling pathway.3 Ras is inactive when attached to GDP and becomes active when attached to GTP. As a negative regulator of the Ras-MAP kinase signaling pathway, the Spred-1 protein inhibits activation of Raf by activating Ras.3
For this reason, Legius syndrome is considered a member of the group known as RASopathies.4 Other syndromes in this group include NF1 and the Noonan, cardio-facio-cutaneous, LEOPARD, Costello, capillary malformation–arteriovenous malformation and CBL mutation-associated syndromes.
The prevalence of Legius syndrome remains unknown, but RASopathies affect 1 in every 1,000 individuals.4 Nearly 200 cases of Legius syndrome have been described in the literature. Familial as well as sporadic cases have been reported.5 The proportion of patients who, like our case patient, have macrocephaly (≥97th percentile) is estimated to be 42%.6
In addition to macrocephaly and CALMs, there are other overlapping clinical manifestations between NF1 and Legius syndrome (see Table), including freckling and possible facial dysmorphy and pulmonary valve stenosis.6

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/8e080468-610e-41a5-8b2f-349c2e719aec/19-NEU-430-Moodley-NF1-Legius-Syndrome_jpg)
Developmental delay has been reported in some published series of Legius syndrome cases, including in 11 of 40 patients described by Messiaen and colleagues.5 Additionally, among 25 Legius syndrome cases described by Denayer et al.,7 five patients presented with motor delay, five with speech delay, three with attention deficit hyperactivity disorder (ADHD) and 14 with learning difficulties. The latter researchers also found cognitive impairment in terms of lower performance IQ among children with Legius syndrome compared with their unaffected family members.7
Advertisement
The case profiled here is the first known case of Legius syndrome reported in Ohio. The impetus for this report is a desire to raise awareness of this syndrome so that clinicians increasingly consider it in their differential diagnosis in appropriate settings, as accurate diagnosis of Legius syndrome has important implications for management, counseling and prenatal diagnosis.
Although the phenotype of Legius syndrome has been described as milder than that of NF1, these findings are based on a small number of cases and very limited clinical data, and the less-common complications associated with Legius syndrome are not completely understood.
Distinguishing between NF1 and Legius syndrome is challenging in children who have multiple CALMs but no other signs of NF1, particularly if the patient is young, because most clinical findings in NF1 develop with age.8 Yet proper diagnostic distinction is important, since medical management differs significantly between the two conditions.
For these reasons, genetic testing can play a very important role, as the diagnosis of NF1 does not require genetic testing,9 whereas genetic testing is necessary for a definitive diagnosis of Legius syndrome. In the case reported here, the need for genetic testing was driven by the paucity of clinical signs for NF1 and by significant parental concern for a definitive diagnosis.
Differences between the conditions extend to management, as prognosis is generally better for Legius syndrome, translating to less-stringent medical follow-up and less psychological burden for families compared with NF1.
Advertisement
Whereas children with known or suspected NF1 typically require regular neurological evaluation (as well as regular follow-up with a pediatric ophthalmologist), regular neurological evaluations are not necessary for those with Legius syndrome. If our patient’s genetic testing had identified a mutation consistent with NF1, he would have been evaluated by a clinical geneticist promptly. However, since his testing for NF1 was normal, he has been recommended for follow up with genetics staff in approximately one year, or sooner if additional concerns arise.
For the associated manifestations of Legius syndrome, the following measures are recommended:
Surveillance for individuals with Legius syndrome is recommended, as is routine screening for developmental delays and behavioral and learning problems.10
The presence of CALMs in a child showing few or no other diagnostic features of NF1 should alert physicians to the possibility of Legius syndrome. However, clinical examination alone is insufficient to make a definitive diagnosis, and genetic testing is necessary. Some developmental delay might be present in a child with Legius syndrome, typically milder than in NF1. Overall, less-intense medical surveillance is required for Legius syndrome than for NF1.
Dr. Moodley is a staff physician in Cleveland Clinic’s Center for Pediatric Neurosciences and co-director of its Neurofibromatosis Program.
Advertisement

Findings may have implications for understanding the disorders’ pathophysiology

Routine capture of standardized neuroperformance data may expand and refine investigations

Nearly one-fifth of such cases fulfill 2024 McDonald criteria based on biomarkers

Adequate dosing may improve outcomes in well-selected patients, large Cleveland Clinic series suggests

Reimagining the outpatient neurological visit with routine capture of neuroperformance data

Free portal helps researchers classify and share data using the IC-CoDE framework

Large study shows rural patients are less apt to be discharged to inpatient rehab, hampering outcomes

Updates on this fast-evolving therapeutic landscape from a leading trialist