Insights into an increasingly recognized cause of encephalitis
By Sumit Parikh, MD, and Trishna Kantamneni, MD
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
A 2.5-year-old previously healthy girl presented to Cleveland Clinic Children’s following a first-time prolonged complex partial seizure with persistent abnormal movements of her extremities.
Over the next few days she developed orofacial dyskinesia, dystonia and choreoathetoid movements of the extremities. She began to lose her speech, had progressive difficulty with swallowing and became increasingly agitated. Her family history was significant for an older brother with a similar presentation of severe encephalopathy, seizures and abnormal movements of unknown etiology around the same age.
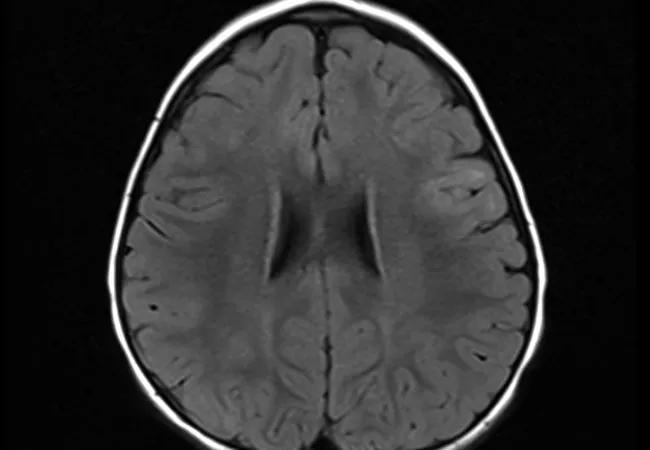
Continuous video EEG to rule out epilepsia partialis continua showed focal slowing on EEG. Brain MRI was significant for subtle FLAIR signal changes in the right frontoparietal region involving the precentral gyrus (Figure).
A lumbar puncture was done to look for underlying inflammatory, metabolic and autoimmune disorders, including paraneoplastic disease. She was started on a high-dose IV steroid infusion in view of the rapid clinical progression and high clinical suspicion for autoimmune encephalitis. CSF showed mild pleocytosis and oligoclonal bands. Her serum and CSF were positive for anti-N-methyl-D-aspartate receptor (NMDAR) antibodies. In the interim, a complete workup looking for an occult tumor was unremarkable.
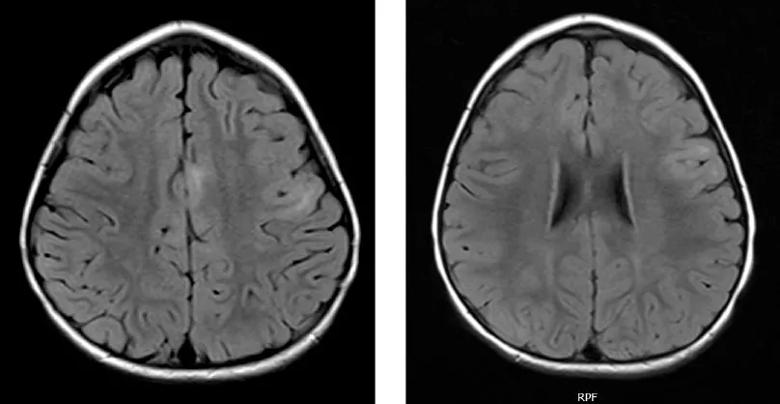
Figure. Axial FLAIR images demonstrating subtle hyperintensities in the left middle and inferior frontal gyrus.
She was started on immune-modulating therapy immediately following the diagnosis. She completed a course of rituximab and cyclophosphamide. She continues on monthly IVIG as long-term mycophenolate therapy has been initiated. Her seizures are well-controlled on topiramate monotherapy. Her agitation and hyperkinetic movements have improved significantly. She is receiving intensive rehabilitation therapies at Cleveland Clinic Children’s Hospital for Rehabilitation.
Anti-NMDAR encephalitis is an increasingly recognized cause of autoimmune encephalitis in children. First described by Dalmau and colleagues in 2007 (see Bibliography below), it is associated with antibodies against the GluN1 subunit of the NMDA receptor. Young children usually present with abnormal movements and seizures and progress to having abnormal behavior and cognition, decreased level of consciousness, speech disorder, autonomic dysfunction, sleep disorders and hypoventilation. It is less frequently associated with an underlying tumor in children (0 to 5 percent in patients < 12 years of age) than in females older than 18 (~58 percent).
Workup should include a lumbar puncture to rule out other causes of acute encephalitis and assess for anti-NMDAR antibodies. There can be pleocytosis and oligoclonal bands in the CSF. Brain MRI and EEG can be normal or show nonspecific changes. Patients should be screened for an occult neoplasm, especially ovarian teratomas.
There are no standard immunotherapy protocols for autoimmune encephalitis. Steroids, IVIG or plasmapheresis and removal of an identifiable tumor are considered first-line treatment. In the absence of clinical response, second-line treatment includes rituximab and/or cyclophosphamide. About 75 percent of patients have full or substantial recovery after immunotherapy or tumor removal. Relapses are reported in about 25 percent of patients.
While anti-NMDAR encephalitis remains the prototypical autoimmune encephalitis, over a dozen new autoantibodies have been identified since the initial description. Some of these syndromes are due to antibodies directed against the neuronal cell surface or synaptic antigens, while others are associated with intracellular synaptic proteins (e.g., paraneoplastic limbic encephalitis). As noted by Leypoldt et al. (see Bibliography below), most neuronal cell surface or synaptic autoantibodies are IgG1 or IgG3 subtypes that potentially fix complement but with no evidence of complement-mediated mechanisms of cell injury to date. This appears different from the aquaporin 4 antibodies in neuromyelitis optica, for example, where symptoms are less reversible and complement-mediated brain injury is more visible on MRI.
Although it remains rare, anti-NMDAR encephalitis has now been reported in a number of series to surpass any individual viral etiology as the cause of autoimmune encephalitis in young children.
Dr. Parikh is a staff physician in Cleveland Clinic’s Center for Pediatric Neurosciences.
Dr. Kantamneni is a pediatric neurology fellow in Cleveland Clinic’s Center for Pediatric Neurosciences.
Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25-36.
Florance NR, Davis RL, Lam C, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in Children and Adolescents. Ann Neurol. 2009;66:11-18.
Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15:391-404.
Leypoldt F, Amangue T, Dalmau J. Autoimmune encephalopathies. Ann N Y Acad Sci. 2015;1338:94-114.
Zekeridou A, Karantoni E, Viaccoz A, et al. Treatment and outcome of children and adolescents with N-methyl-D-aspartate receptor encephalitis. J Neurol. 2015;262:1859-1866.
When specialized surgery makes sense for moyamoya syndrome
Multilevel cervical fusion restores function in an athletic 78-year-old
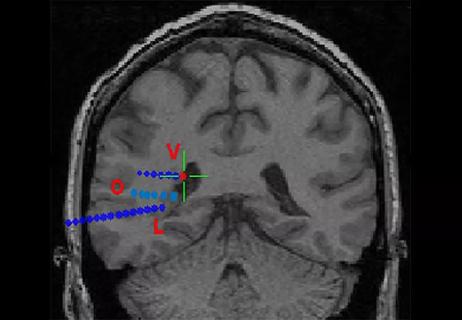
Case study underscores the imperative for thorough evaluation with SEEG
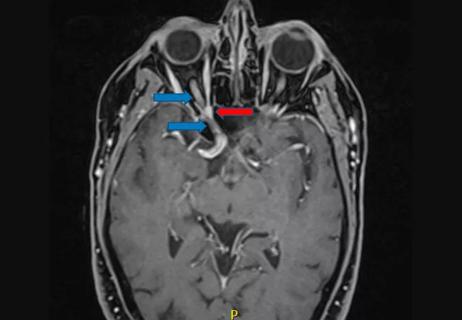
Schwannoma of the lacrimal nerve threatened right eye blindness
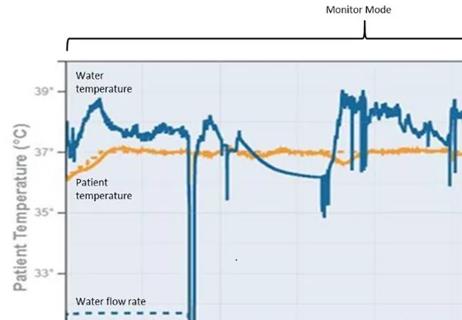
Case report demonstrates utility in a brain-injured patient
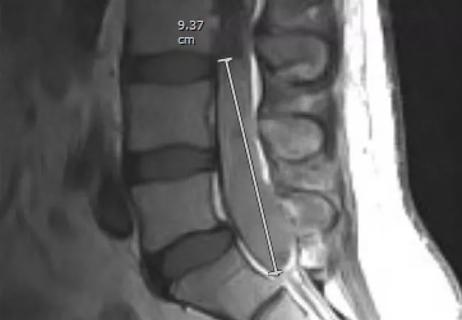
Partial resection plus radiation leads to good outcome from an unpredictable tumor
Diagnosis and treatment of rotational vertebrobasilar insufficiency syndrome
First reported case expands use of minimally invasive techniques