Locations:

Holistic approach is necessary to ensure a correct diagnosis
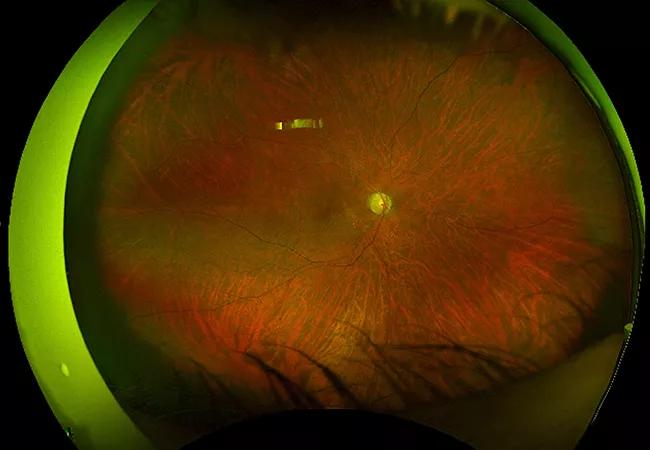
Above: An ophthalmologic exam with fluorescence angiogram was notable for severe occlusive vasculitis and posterior uveitis.
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
By Rula A. Hajj-Ali, MD, Komal Ejaz, MD, and Jeffrey Donaldson, MD
Central nervous system vasculitis (CNSV) is fraught with diagnostic challenges. Clinical presentation can be quite variable, and there is no disease-specific test. Furthermore, the condition has several mimics, even with the availability of brain tissue. Clinicians should always be on alert for other diagnostic possibilities that could present as CNSV.
Here we present a case in which the diagnosis of CNSV was challenged.
A 50-year-old male was referred to Cleveland Clinic’s Center for Vasculitis Care and Research for evaluation of four years of neurological symptoms. His initial presentation included intermittent headaches, forgetfulness, and cognitive and personality changes, with right upper and lower extremity weakness, and loss of sensation.
The initial work-up revealed multifocal regions of parenchymal signal abnormality involving the left parietal lobe, frontal lobe, and corpus callosum with scattered areas of restricted diffusion on magnetic resonance imaging (MRI) of the brain. Cerebrospinal fluid analysis revealed an elevated protein of 233 mg/mL with a white cell count of 34/mm3. A fluorescein angiogram (FA) performed for visual changes revealed retinal vasculitis. A brain biopsy showed small vessel vasculopathy with coagulative type necrosis.
The patient was diagnosed with primary CNSV and treated with high-dose glucocorticoids and oral cyclophosphamide for six months, followed by mycophenolate for maintenance. Despite aggressive treatment measures, his disease continued to progress, with worsening motor weakness and sensory and personality changes.
Advertisement
Serial MRI imaging revealed new lesions and a worsening of existing lesions. He was then treated with one course of rituximab. However, he continued to have clinical and radiographic progression and was subjected to another course of oral cyclophosphamide for six months; mycophenolate was continued.
Unfortunately, he developed complications, including pleural regenerative hyperplasia); hypercalcemia; anemia requiring multiple blood transfusions; bilateral extensive deep venous thromboses requiring anticoagulants and, finally, worsening kidney disease.
At our Center for Vasculitis Care and Research, we take a systematic approach in the work-up of any patient suspected of having CNSV. This includes a thorough review of symptoms and radiologic and laboratory findings with detailed evaluation for mimics, including infectious, genetic and malignant causes. It is vital to delineate details of any therapeutic trials and available tissue biopsies.
This case raised multiple flags that called for reconsideration of the diagnosis, including lack of response to multiple rounds of effective treatment, findings of retinal vascular disease and systemic involvement. The differential diagnosis of conditions that affect the brain and the eye includes neurosarcoidosis, Susacs syndrome, demyelinating diseases and lymphoma.
The patient lacked specific manifestations of neurosarcoidosis or granulomatous inflammation on biopsy and had a negative positron emission tomography scan. Multiple sclerosis was considered, but the patient lacked the characteristic clinical course and consistent MRI findings.
Advertisement
Refractoriness to adequately dosed immunosuppression, and accompanying liver and kidney disease with anemia, raised suspicion for retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations (RVCL-S) caused by TREX1 mutation. Prior brain pathology revealed small vessel vasculopathy with coagulative necrosis, which has been reported in RVCL-S. Magnetic resonance angiography and 7-Tesla MRI of the brain were obtained along with ophthalmology evaluation. The ophthalmologic exam was notable for bilateral severe occlusive vasculitis and posterior uveitis, with FA showing significant disc and large vessel leakage.
This patient did not have a family history to support the typical autosomal dominant inheritance pattern of TREX1 mutation, but a de novo mutation was suspected. De novo mutations, while rare, have been described as heterozygous frameshift mutations involving TREX1. Subsequent genetic testing performed after genetic counseling revealed TREX1 C-terminal frameshift mutation with a likely pathogenic variant.
RVCL-S is an exceedingly rare and uniformly fatal genetic disease that affects the microvasculature primarily of the brain and eye. The onset of this disease is usually reported between 35 and 50 years. This genetic disease is caused by somatic or, rarely, de novo mutations of the TREX1 gene, which is involved in DNA repair.
RVCL-S is characterized by visual field defects caused by vascular retinopathy as well as a series of micro-infarcts in the brain leading to encephalopathy, focal neurological signs, or global brain dysfunction leading to cognitive decline. Blood vasculature in other organs can be impacted, leading to liver and renal involvement, anemia, hypertension, thyroid disease, migraines, osteonecrosis, intestinal bleeding and Raynaud’s phenomenon. Unfortunately, life expectancy following symptom onset is reduced to five to 20 years.
Advertisement
Imaging findings of RVCL-S include supratentorial lesions, brain calcifications and cerebral atrophy. Lesions can display surrounding edema and mass effect. Cerebellar punctate-enhancing lesions are typically seen in patients older than 50.
While there is currently no treatment for RVCL, multiple investigations are underway. An ongoing phase 2 trial is looking at the efficacy and safety of crizanlizumab, a humanized monoclonal anti-P-selectin antibody that prevents leukocyte adhesion to the vascular endothelium, thereby limiting the risk of microvascular occlusion and leading to fewer ischemic brain and eye lesions.
Furthermore, researchers at the University of Pennsylvania are investigating the role of delivering therapies to correct the genetic mutation in RVCL, which may prove to be a breakthrough in the near future.
1. de Boer, I., Pelzer, N., & Terwindt, G. (2019). Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. GeneReviews®
2. Grand MG, Kaine J, Fulling K, et al. Cerebroretinal vasculopathy: A new hereditary syndrome. Ophthalmology.1988;95:649-653.
3. Gutmann DH, Fischbeck KH, Sergott RC. Hereditary retinal vasculopathy with cerebral white matter lesions. Am J Med Genet.1989;34:217-220.
4. Stam AH, Kothari PH, Shaikh A, et al. Retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations. Brain. 2016;139:2909-2922.
Advertisement
Advertisement

Evidence-based therapies, monitoring, prevention and more
Knowing the affected organs and vessels can help in identifying cause

Unraveling the TNFA receptor 2/dendritic cell axis

Nasal bridge inflammation, ear swelling and neck stiffness narrow the differential diagnosis

Genetic testing at Cleveland Clinic provided patient with an updated diagnosis
Combining quantitative vessel wall MRI metrics, CSF abnormalities and neurologic symptoms can be highly predictive

From dryness to diagnosis

Multiple comorbidities are associated with pediatric psoriasis