Locations:

Good visual recovery and retinal reperfusion after prompt diagnosis and intensive therapy
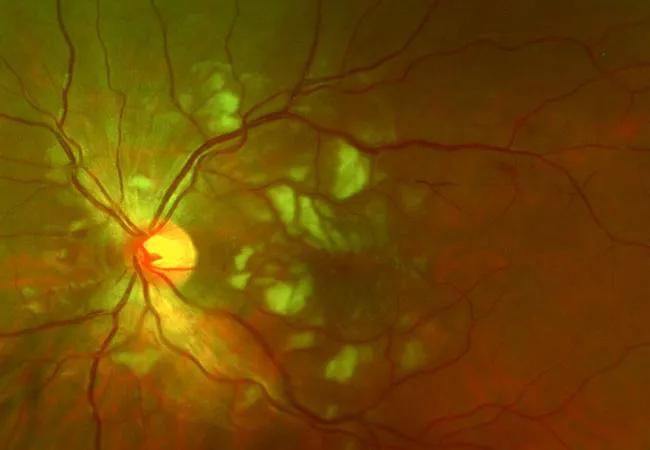
Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/a7603c31-a4e6-443a-bade-b89a49531be3/650x450-Zeft_jpg)
Vaso-occlusive retinopathy
by Andrew Zeft, MD, MPH, and Careen Lowder, MD, PhD
Advertisement
Cleveland Clinic is a non-profit academic medical center. Advertising on our site helps support our mission. We do not endorse non-Cleveland Clinic products or services. Policy
A 15-year-old boy presented with a 2-week history of sudden, blurred vision in the left eye. His past medical history was significant for a diagnosis of systemic lupus erythematosus (SLE) made one year previously by pediatric rheumatology.
At the time of SLE diagnosis, he developed a malar rash and was found to have elevated titers of anti-double stranded DNA antibodies (312, nl < 30 IU/mL), anti-Smith antibodies (6.1, nl < 1.0 AI), anti-ribonucleoproteins antibodies (4.2, nl < 1.0 AI), Telanti-Sjogren’s syndrome antigen A antibodies (8.0, nl < 1.0 AI), anti-nuclear antibody (speckled pattern, 1:1280), Crithidia lucillae testing positive, and low complement C4 levels (6, nl 12–46 mg/dL). He maintained good control of his symptoms on hydroxychloroquine (400 mg once daily) and prednisone (5 mg once daily).
On incremental workup by rheumatology, he was found to have an elevated anti-cardiolipin IgM antibody (32, nl 0–11 MPL) and anti-beta 2 glycoprotein IgM antibody (70, nl < 20 SMU) titers indicative of possible anti-phospholipid syndrome (APLS). His anti-cardiolipin IgM antibody (33 MPL) and anti-beta 2 glycoprotein IgM antibody (52 MPL) titers remained elevated four months later on confirmatory testing, thus meeting criteria for a clinically significant antiphospholipid profile. He did not have any clinical signs of vascular thrombosis and he did not have any other clinical manifestations of SLE beyond a malar rash. He was started on prophylactic aspirin 81 mg once daily.
At the time of first ocular examination, visual acuity (VA) was 20/20 in the right eye and 20/80 in the left eye. There was no relative afferent pupillary defect. Intraocular pressure, confrontational visual fields and extraocular movements were normal. Anterior segment examination was normal. There were no signs of anterior vitreous inflammation. The right fundus was normal. Fundus examination of the left eye demonstrated multiple clustered cotton wool spots in the macula and posterior pole. In the retina periphery, there was segmental sheathing of both arterioles and venules. On optical coherence tomography (OCT), there was inner retinal hyperreflectivity and thickening, cystoid macular edema and subretinal fluid. Intravenous fluorescein angiography (IVFA) was normal in the right eye, but the left eye showed marked macular capillary non-perfusion, arteriole occlusions, delayed venous filling, late macular leakage and segmental peripheral vascular leakage. Optical coherence tomography angiography (OCTA) revealed significant flow void in the macula of the left eye.
Advertisement

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/9ea51dcb-260e-4b81-b980-2f8b3ccde7aa/fundus-photo-cotton-wool-spots-macula)
Fundus photograph of the left eye at the time of patient presentation showing numerous cotton wools spots in the macula and posterior pole.

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/e47dd5b4-55dd-4ab3-ba5a-a5c087744238/angiography-retinal-artery-occlusion)
Late fluorescein angiogram of the left eye demonstrating multiple branch retinal artery occlusions, diffuse vascular leakage in the macula, and macular capillary non-perfusion.
In the setting of new vasothrombotic disease and elevated anti-phospholipid antibody titers, a diagnosis of vaso-occlusive retinopathy from APLS was made. The patient was promptly treated with intravenous (IV) methylprednisolone 1 g once daily x 3 days followed by systemic prednisone at 1 mg/kg daily (60 mg once daily) and tapered by 5 mg every week as able. The patient was simultaneously started on mycophenolate mofetil (1000 mg two times per day). At the time of vision loss, his APLS panel remained elevated with an anti-cardiolipin IgM of 20 MPL and an anti-beta2-glycoprotein IgM of 25 SMU. His lupus laboratory tests were abnormal with a low C4 level (9 mg/dL) and an elevated anti-dsDNA antibody titer (312 IU/mL) indicating disease activity.
Despite high-dose IV steroids there was minimal improvement detected on interval follow-up OCTA and IVFA. Without improvement in retinal perfusion there was significant concern that permanent visual loss was imminent. One week after initial exam and further discussion with our pediatric rheumatology and hematology/oncology colleagues regarding the risks and benefits of full anti-coagulation in an active 15-year-old boy, the decision was made to therapeutically anti-coagulate the patient with enoxaparin (1 mg/kg every 12 hours, titrated by anti-factor Xa level).
The patient was followed closely and there was remarkable interval improvement in clinical exam, OCTA retinal capillary flow and IVFA vascular leakage. At three months follow-up, the patient’s VA improved to 20/20 in the affected left eye. OCTA demonstrated reconstitution of flow in the retinal capillary plexus. In addition, there was resolution of cystoid macular edema and subretinal fluid on OCT and reperfusion of previously occluded arterioles on IVFA. Anti-cardiolipin antibody and anti-beta 2 glycoprotein antibody titers normalized. At four-month follow-up, there was resolution of all cotton wool spots on fundus exam. However, there were new areas of vascular leakage on IVFA as his oral prednisone was tapered to 15 mg once daily while on mycophenolate 1500 mg twice daily. Rituximab infusions (1000 mg) were initiated at this follow-up visit with a second infusion two weeks later in attempt to taper the patient off systemic steroids. The patient was successfully tapered off systemic steroids by month six of follow-up. At 10-month follow-up, the patient showed complete resolution of vascular leakage on IVFA. On final follow-up, 20 months after initial presentation, the patient’s VA remained at 20/20 in both eyes on a regimen of aspirin 81 mg once daily, mycophenolate mofetil 1500 mg twice daily, and off of systemic steroids and anti-coagulation therapy. Anti-cardiolipin antibody, anti-beta 2 glycoprotein, anti-Xa activity remained negative.
Advertisement

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/5d964e91-526b-46d0-b01c-3234f9176852/fundus-photo-healthy-macula)
Fundus photo of the left eye at 4-month follow-up showing resolution of cotton wool spots.

Image content: This image is available to view online.
View image online (https://assets.clevelandclinic.org/transform/04f03b04-d370-4341-aa0d-754395ffef6f/angiography-complete-resolution-vascular-leakage)
Late fluorescein angiography of the left eye at 10-month follow-up demonstrating complete resolution of vascular leakage.
SLE is a protean autoimmune connective tissue disorder that can involve nearly every organ system, and affected patients can present with an array of clinical manifestations.1 SLE can affect any part of the eye including the orbit, eyelids, conjunctiva, cornea, sclera, retina, choroid and optic nerve.1-3 APLS is a related autoimmune disorder characterized clinically by recurrent vascular thromboembolic events and fetal loss.4 APLS can occur independently of other autoimmune disorders, but is most commonly associated with SLE, occurring in up to 30% of patients with previously diagnosed lupus.5 The diagnosis of APLS is made based upon the presence of clinical vascular thrombosis and serological presence of elevated anti-cardiolipin antibodies or anti-beta2-glycoprotein titers.4 Vaso-occlusive retinopathy from APLS is a rare form of retinopathy in patients with SLE, but its visual prognosis is reportedly grim.6,7
In this case, the patient was treated with intravenous methylprednisolone, mycophenolate motefil, aspirin and enoxaparin. The patient not only had great recovery of visual acuity, but also demonstrated reperfusion of arterioles and reconstitution of flow in the retinal capillary network. These findings suggest that the vaso-occlusive disease is reversible if the diagnosis is made promptly and intensive therapy is initiated.
Advertisement
*Note: This is an abridged version of an article previously published by the American Journal of Ophthalmology Case Reports, under a Creative Commons license.
Advertisement
Advertisement
How to use? Consider starting during the acute attack and seek patient preferences for chronic use

Distinct MRI signature includes lesions beyond the corpus callosum, features predictive of vision and hearing loss

Despite less overall volume loss than in MS and NMOSD, volumetric changes correlate with functional decline

It’s time to get familiar with this emerging demyelinating disorder
Early experience with the agents confirms findings from clinical trials
Despite advancements, care for this rare autoimmune disease is too complex to go it alone
Treatment for scleroderma can sometimes cause esophageal symptoms
Patient’s symptoms included periorbital edema, fevers, weight loss